Reproduce results in Figure 4
Related dataset:
Reference of breast cancer can be downloaded here
All the datasets are training with epoch settings: epochs = 200 seg_training_epochs = 10 deconv_warmup_epochs = 100
All the data for reproducing the result can be downloaded from Zenodo. The data used in this notebook is under the folder figure_4.
[1]:
import os
# Download the data from the link above to this folder and unzip it, can be changed to your own path
os.chdir("/import/home3/yhchenmath/Dataset/CellARTPaper/figure_4/")
CellART annotation on two replicates
[2]:
import numpy as np
import scanpy as sc
import pandas as pd
import matplotlib.pyplot as plt
import tifffile
import spatialdata_plot
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/numba/core/decorators.py:282: RuntimeWarning: nopython is set for njit and is ignored
warnings.warn('nopython is set for njit and is ignored', RuntimeWarning)
[3]:
celltype_mapping = {
'B Cells': '#1E77B4',
'DCs': '#E477C1',
'Endothelial': '#289E68',
'Macrophages': '#D62728',
'Malignant': '#FF7F0D',
'Mast Cells': '#8C564B',
'Myoepithelial': '#AA41FC',
'Perivascular-Like': '#B6BD61',
'Stromal': '#15BECF',
'T Cells': '#AFC7E8',
}
[4]:
from matplotlib.gridspec import GridSpec
from matplotlib.lines import Line2D
annotated_svt = sc.read_h5ad("adata_breast_cancer_rep1.h5ad")
df = annotated_svt.obs
fig, ax1 = plt.subplots(1, 1, figsize=(8, 6))
from matplotlib.patches import Rectangle
celltype_names = list(celltype_mapping.keys())
selected_celltype = celltype_names
for i in range(len(celltype_names)):
if celltype_names[i] not in selected_celltype:
continue
sub_df = df[df["celltype"] == celltype_names[i]]
ax1.scatter(sub_df["y"], sub_df["x"], s=0.05, label=celltype_names[i], color=celltype_mapping[celltype_names[i]])
# ax1.invert_yaxis()
ax1.axis("off")
ax1.set_xlim(df["y"].min(), df["y"].max())
ax1.set_ylim(df["x"].max(), df["x"].min())
ax1.invert_yaxis()
plt.show()

[5]:
annotated_svt = sc.read_h5ad("adata_breast_cancer_rep2.h5ad")
df = annotated_svt.obs
fig, ax1 = plt.subplots(1, 1, figsize=(8, 6))
celltype_names = list(celltype_mapping.keys())
selected_celltype = celltype_names
for i in range(len(celltype_names)):
if celltype_names[i] not in selected_celltype:
continue
sub_df = df[df["celltype"] == celltype_names[i]]
ax1.scatter(sub_df["y"], sub_df["x"], s=0.05, label=celltype_names[i], color=celltype_mapping[celltype_names[i]])
ax1.axis("off")
ax1.set_xlim(df["y"].min(), df["y"].max())
ax1.set_ylim(df["x"].max(), df["x"].min())
ax1.invert_yaxis()
plt.show()
Segmentation and annotation comparison
[6]:
from spatialdata_io import xenium
sdata = xenium(
path="/import/home2/yhchenmath/Dataset/CellSeg/TestSeg/Xenium-BreastCancer1/outs/")
INFO reading /import/home2/yhchenmath/Dataset/CellSeg/TestSeg/Xenium-BreastCancer1/outs/cell_feature_matrix.h5
/tmp/ipykernel_3539281/540348584.py:3: DeprecationWarning: The default value of `cells_as_circles` will change to `False` in the next release. Please pass `True` explicitly to maintain the current behavior.
sdata = xenium(
INFO Transposing `data` of type: <class 'dask.array.core.Array'> to ('c', 'y', 'x').
[25]:
method_colors = {
"CellART": "#B07AA1",
"BIDCell": "#568F18",
"10X": "#7EA5C8",
"Baysor": "#F4B860",
"Cellpose": "#CABFAB"
}
celltype_mapping = {
'B Cells': '#1E77B4',
'DCs': '#E477C1',
'Endothelial': '#289E68',
'Macrophages': '#D62728',
'Malignant': '#FF7F0D',
'Mast Cells': '#8C564B',
'Myoepithelial': '#AA41FC',
'Perivascular-Like': '#B6BD61',
'Stromal': '#15BECF',
'T Cells': '#AFC7E8',
}
cellart_mask = np.load("cellart_segmentation_mask.npy").astype("int32")
cellart_nuclei = np.load("nuclei_segmentation_mask.npy") # U
bidcell_mask = tifffile.imread("bidcell_segmentation_mask.tif")
xenium_mask = tifffile.imread("10X_cell_segmentation_mask.tif")
baysor_mask = tifffile.imread("baysor_segmentation_mask.tif")
cellpose_mask = tifffile.imread("cellpose_nuclei.tif")
annotated_cellart = sc.read_h5ad("adata_breast_cancer_rep1.h5ad")
annotated_10X_scvi = sc.read_h5ad("10X_scvi.h5ad")
annotated_10X_scvi.obs["celltype"] = annotated_10X_scvi.obs["C_scANVI"]
annotated_10X_scvi.obs["center_x"] = annotated_10X_scvi.obsm["spatial"][:, 0]
annotated_10X_scvi.obs["center_y"] = annotated_10X_scvi.obsm["spatial"][:, 1]
annotated_10X_tangram = sc.read_h5ad("10X_tangram.h5ad")
annotated_10X_tangram.obs["celltype"] = annotated_10X_tangram.obsm["tangram_ct_pred"].idxmax(axis=1)
annotated_10X_tangram.obs["celltype"] = annotated_10X_tangram.obs["celltype"].astype("category")
annotated_10X_tangram = annotated_10X_tangram[:, 1:]
annotated_10X_tangram.obs_names = annotated_10X_scvi.obs_names
annotated_10X_rctd = sc.read_h5ad("10X_rctd.h5ad")
[ ]:
# nuclei_cellart = sc.read_h5ad("/import/home2/yhchenmath/Code/Triplet/LOG/XeniumBreastCancer_Annotation_with_feature/epoch_100/cell_deconv.h5ad")
[ ]:
from cellart.utils.spatialdata_utils import append_xenium_boundary
[10]:
append_xenium_boundary(cellart_mask, sdata, "cellart_boundaries", celltype = annotated_cellart.obs["celltype"])
append_xenium_boundary(cellart_nuclei, sdata, "cellart_nuclei")
append_xenium_boundary(bidcell_mask, sdata, "bidcell_boundaries")
append_xenium_boundary(xenium_mask, sdata, "10X_boundaries")
append_xenium_boundary(baysor_mask, sdata, "baysor_boundaries")
append_xenium_boundary(cellpose_mask, sdata, "cellpose_boundaries")
append_xenium_boundary(xenium_mask, sdata, "10X_tangram", celltype = annotated_10X_tangram.obs["celltype"])
append_xenium_boundary(xenium_mask, sdata, "10X_scvi", celltype = annotated_10X_scvi.obs["celltype"])
append_xenium_boundary(xenium_mask, sdata, "10X_rctd", celltype = annotated_10X_rctd.obs["celltype"])
/tmp/ipykernel_3539281/2510591147.py:92: UserWarning: Geometry is in a geographic CRS. Results from 'centroid' are likely incorrect. Use 'GeoSeries.to_crs()' to re-project geometries to a projected CRS before this operation.
center_df = sdata.shapes[append_name].centroid
/tmp/ipykernel_3539281/2510591147.py:92: UserWarning: Geometry is in a geographic CRS. Results from 'centroid' are likely incorrect. Use 'GeoSeries.to_crs()' to re-project geometries to a projected CRS before this operation.
center_df = sdata.shapes[append_name].centroid
/tmp/ipykernel_3539281/2510591147.py:92: UserWarning: Geometry is in a geographic CRS. Results from 'centroid' are likely incorrect. Use 'GeoSeries.to_crs()' to re-project geometries to a projected CRS before this operation.
center_df = sdata.shapes[append_name].centroid
/tmp/ipykernel_3539281/2510591147.py:92: UserWarning: Geometry is in a geographic CRS. Results from 'centroid' are likely incorrect. Use 'GeoSeries.to_crs()' to re-project geometries to a projected CRS before this operation.
center_df = sdata.shapes[append_name].centroid
[11]:
import spatialdata as sd
seg_transformation = sd.transformations.get_transformation(sdata.shapes["nucleus_boundaries"])
translation = sd.transformations.Translation([6, 6.5], axes=("x", "y"))
sequence = sd.transformations.Sequence([translation, seg_transformation])
sd.transformations.set_transformation(sdata.shapes["cellpose_boundaries"], sequence, to_coordinate_system="global")
sd.transformations.set_transformation(sdata.shapes["cellart_boundaries"], sequence, to_coordinate_system="global")
sd.transformations.set_transformation(sdata.shapes["cellart_nuclei"], sequence, to_coordinate_system="global")
sd.transformations.set_transformation(sdata.shapes["bidcell_boundaries"], sequence, to_coordinate_system="global")
sd.transformations.set_transformation(sdata.shapes["baysor_boundaries"], sequence, to_coordinate_system="global")
sd.transformations.set_transformation(sdata.shapes["10X_tangram"], sequence, to_coordinate_system="global")
sd.transformations.set_transformation(sdata.shapes["10X_scvi"], sequence, to_coordinate_system="global")
sd.transformations.set_transformation(sdata.shapes["10X_rctd"], sequence, to_coordinate_system="global")
[12]:
x_min, x_max, y_min, y_max = 12875, 13075, 26100, 26300
sdata_roi_1 = sdata.query.bounding_box(
min_coordinate=[x_min, y_min], max_coordinate=[x_max, y_max], axes=("y", "x"), target_coordinate_system="global"
)
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/anndata/_core/anndata.py:183: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
[13]:
def plot_seg(sdata, img_key, shape_key, label, ax, color, channel = None):
tmp = sdata.pl.render_images(img_key, channel=channel)
tmp = tmp.pl.render_shapes(
shape_key,
color=color,
fill_alpha=0.4,
outline_color=color,
outline_alpha=1,
outline_width=2,
).pl.show(title='', frameon=False, legend_loc='none', ax=ax, return_ax=True, colorbar=False)
ax.axis('off')
[16]:
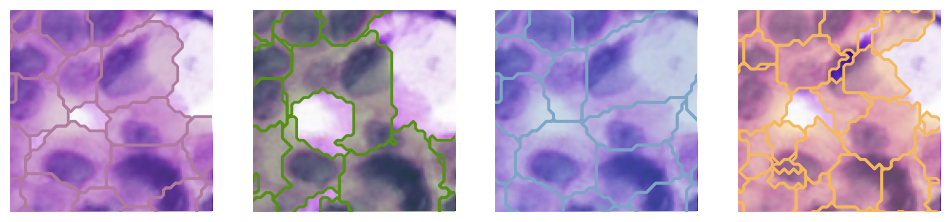
fig, ax = plt.subplots(1, 4, figsize=(12,3))
plot_seg(sdata_roi_1, "he_image", "cellart_boundaries", "CellART", ax[0], method_colors["CellART"])
plot_seg(sdata_roi_1, "he_image", "bidcell_boundaries", "BIDCell", ax[1], method_colors["BIDCell"])
plot_seg(sdata_roi_1, "he_image", "10X_scvi", "10X", ax[2], method_colors["10X"])
plot_seg(sdata_roi_1, "he_image", "baysor_boundaries", "Baysor", ax[3], method_colors["Baysor"])
for i in range(4):
ax[i].set_xlim(y_min, y_max)
ax[i].set_ylim(x_min, x_max)
INFO Value for parameter 'color' appears to be a color, using it as such.
Clipping input data to the valid range for imshow with RGB data ([0..1] for floats or [0..255] for integers).
INFO Value for parameter 'color' appears to be a color, using it as such.
Clipping input data to the valid range for imshow with RGB data ([0..1] for floats or [0..255] for integers).
INFO Value for parameter 'color' appears to be a color, using it as such.
Clipping input data to the valid range for imshow with RGB data ([0..1] for floats or [0..255] for integers).
INFO Value for parameter 'color' appears to be a color, using it as such.
Clipping input data to the valid range for imshow with RGB data ([0..1] for floats or [0..255] for integers).
[17]:
x_min, x_max, y_min, y_max = 16750, 19750, 17750, 20750
sdata_roi_2 = sdata.query.bounding_box(
# 3500, 4200, 3700, 4500
min_coordinate=[16750, 17750], max_coordinate=[19750, 20750], axes=("y", "x"), target_coordinate_system="global"
)
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/anndata/_core/anndata.py:183: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
[18]:
for k in ["cellart_boundaries", "10X_tangram", "10X_scvi", "10X_rctd"]:
ct_col = sdata_roi_2.shapes[k].celltype
cts = sdata_roi_2.shapes[k].celltype.unique()
for ct in cts:
sdata_roi_2.shapes[f"{k}_{ct.replace(' ', '_').replace('(', '').replace(')', '')}"] = \
sdata_roi_2.shapes[k][ct_col == ct]
[19]:
def plot_annotation(sdata, shape_key, ax, title):
tmp = sdata.pl.render_images("he_image")
draw_cts = sdata.shapes[shape_key].celltype.unique().tolist()
for ct in draw_cts:
color = celltype_mapping[ct]
tmp = tmp.pl.render_shapes(
f"{shape_key}_{ct.replace(' ', '_').replace('(', '').replace(')', '')}",
color=color, fill_alpha=0.45, outline_width=0.3, outline_alpha=1, outline_color = color
)
tmp.pl.show(coordinate_systems="global", ax=ax, title="", frameon=False, legend_loc='none')
ax.axis('off')
# ms = 20
# legend_elements = [Line2D([0], [0], marker= None, color='w', label=title, linewidth=0,
# markeredgecolor="#FFFFFF", markerfacecolor="#FFFFFF", markersize=ms)]
# ax.legend(handles=legend_elements, bbox_to_anchor=(0.45,-0.13), loc='lower center', ncol=2, handletextpad=0., prop={'size': 20, 'style': 'italic'}, frameon=False)
[20]:
# plt.style.use('default')
fig, ax = plt.subplots(1, 4, figsize=(16,4))
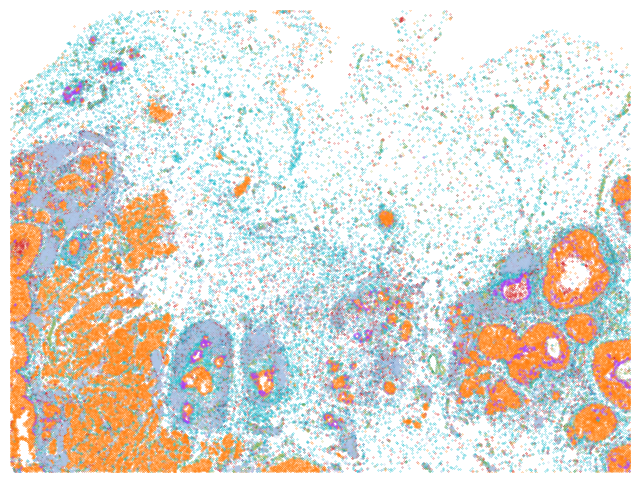
plot_annotation(sdata_roi_2, "cellart_boundaries", ax[1], "CellART (Corr 0.81)")
plot_annotation(sdata_roi_2, "10X_tangram", ax[2], "Xenium 1.0 + Tangram (Corr 0.77)")
plot_annotation(sdata_roi_2, "10X_scvi", ax[3], "Xenium 1.0 + scVI (Corr 0.73)")
g = "CDH1"
sdata_roi_2.shapes["cellart_boundaries"][g] = annotated_cellart[list(sdata_roi_2.shapes["cellart_boundaries"].index.astype(str))].X[:, annotated_cellart.var_names.get_loc(g)].toarray()
sdata_roi_2.pl.render_images(
"he_image").pl.render_shapes(
f"cellart_boundaries",
color=g, fill_alpha=0.8, outline_width=0.3, outline_alpha=1, outline_color = "w", cmap='Oranges'
).pl.show(coordinate_systems="global", ax=ax[0], title="", frameon=False, legend_loc='none', colorbar=False)
ax[0].axis('off')
for i in range(4):
ax[i].set_xlim(y_min, y_max)
ax[i].set_ylim(x_min, x_max)
# plt.tight_layout()
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
Clipping input data to the valid range for imshow with RGB data ([0..1] for floats or [0..255] for integers).
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
Clipping input data to the valid range for imshow with RGB data ([0..1] for floats or [0..255] for integers).
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
Clipping input data to the valid range for imshow with RGB data ([0..1] for floats or [0..255] for integers).
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/geopandas/geodataframe.py:1819: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame.
Try using .loc[row_indexer,col_indexer] = value instead
See the caveats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#returning-a-view-versus-a-copy
super().__setitem__(key, value)
Clipping input data to the valid range for imshow with RGB data ([0..1] for floats or [0..255] for integers).

[21]:
import numpy as np
import seaborn as sns
import matplotlib.pyplot as plt
from matplotlib import rcParams
def plot_bar(df, x_col, y_col, hue, y_label, ax, method_colors, error_bar=False):
# Plot
if error_bar:
sns.barplot(
data=df,
x=x_col,
y=y_col,
hue=hue,
estimator=np.median,
errorbar=('pi', 10),
capsize=0.1,
palette=method_colors,
ax=ax,
err_kws={"linewidth": 1}
)
else:
sns.barplot(
data=df,
x=x_col,
y=y_col,
hue=hue,
capsize=0.1,
palette=method_colors,
ax=ax,
err_kws={"linewidth": 1}
)
# Axis labels and limits
ax.set_ylabel(y_label, fontsize=15)
ax.set_xlabel('')
# ax.set(ylim=(0.18, 0.22))
# Rotate x-tick labels
ax.set_xticklabels(ax.get_xticklabels(), rotation=45, ha='right')
# Remove grid
ax.grid(False)
# Customize spines (axis lines)
ax.spines['left'].set_visible(True)
ax.spines['bottom'].set_visible(True)
ax.spines['left'].set_color('black')
ax.spines['bottom'].set_color('black')
ax.spines['left'].set_linewidth(1)
ax.spines['bottom'].set_linewidth(1)
ax.spines['right'].set_visible(False)
ax.spines['top'].set_visible(False)
# Add small outward ticks
ax.tick_params(
axis='both',
which='major',
direction='out',
length=4,
width=1,
color='black',
bottom=True, top=False, left=True, right=False
)
[28]:
import anndata as ad
adata_sc = sc.read_h5ad("filtered_sc.h5ad")
adata_sc.obs['celltype'] = adata_sc.obs['celltype'].replace({"Invasive Tumor": "Malignant", "DCIS 2": "Malignant", "Prolif Invasive Tumor": "Malignant", "DCIS 1": "Malignant", "CD4+ T Cells": "T Cells", "CD8+ T Cells": "T Cells", "Macrophages 1": "Macrophages", "Macrophages 2": "Macrophages", "Myoepi ACTA2+": "Myoepithelial", "Myoepi KRT15+": "Myoepithelial", "IRF7+ DCs": "DCs", "LAMP3+ DCs": "DCs"})
def find_markers(
adata: ad.AnnData,
cell_type_column: str,
pos_percentile: float = 5,
neg_percentile: float = 10,
percentage: float = 50,
):
"""
Identify positive and negative markers for each cell type based on gene expression.
Parameters:
----------
adata : AnnData
Annotated data object containing gene expression data.
cell_type_column : str
Column name in `adata.obs` specifying cell types.
pos_percentile : float, default=5
Top x% of highly expressed genes to consider as markers.
neg_percentile : float, default=10
Bottom x% of lowly expressed genes to consider.
percentage : float, default=50
Minimum percentage of cells within a type that must express a marker.
Returns:
-------
Dict[str, Dict[str, List[str]]]
Dictionary mapping cell types to lists of positive and negative marker genes.
"""
markers = {}
adata.raw = adata # Ensure raw expression values are used
adata.var_names_make_unique()
sc.tl.rank_genes_groups(adata, groupby=cell_type_column)
genes = adata.var_names
for cell_type in adata.obs[cell_type_column].unique():
subset = adata[adata.obs[cell_type_column] == cell_type]
mean_expression = np.asarray(subset.X.mean(axis=0)).flatten()
# Compute percentile cutoffs
cutoff_high = np.percentile(mean_expression, 100 - pos_percentile)
cutoff_low = np.percentile(mean_expression, neg_percentile)
pos_indices = np.where(mean_expression >= cutoff_high)[0]
neg_indices = np.where(mean_expression <= cutoff_low)[0]
# Filter positive markers based on expression percentage
expr_frac = np.asarray((subset.X[:, pos_indices] > 0).mean(axis=0)).flatten()
valid_pos_indices = pos_indices[expr_frac >= (percentage / 100)]
markers[cell_type] = {
"positive": genes[valid_pos_indices].tolist(),
"negative": genes[neg_indices].tolist(),
}
return markers
purified_markers = find_markers(adata_sc, "celltype", neg_percentile=10, percentage=50, pos_percentile=90)
def calculate_sensitivity(
adata: ad.AnnData, purified_markers, max_cells_per_type: int = 1000
):
"""
Calculate sensitivity of purified markers for each cell type.
Parameters:
----------
adata : AnnData
Annotated data object containing gene expression data.
purified_markers : Dict[str, Dict[str, List[str]]]
Dictionary mapping cell types to positive and negative markers.
max_cells_per_type : int, default=1000
Maximum number of cells to consider per cell type.
Returns:
-------
Dict[str, List[float]]
Sensitivity values for each cell type.
"""
sensitivity_results = {cell_type: [] for cell_type in purified_markers.keys()}
for cell_type, markers in purified_markers.items():
positive_markers = markers["positive"]
subset = adata[adata.obs["celltype"] == cell_type]
if subset.n_obs > max_cells_per_type:
cell_indices = np.random.choice(subset.n_obs, max_cells_per_type, replace=False)
subset = subset[cell_indices]
for cell_counts in subset.X:
positive_indices = subset.var_names.get_indexer(positive_markers)
total_counts = cell_counts.sum()
positive_counts = cell_counts[positive_indices].sum() if positive_markers else 0
sensitivity = positive_counts / total_counts if total_counts > 0 else 0
sensitivity_results[cell_type].append(sensitivity)
return sensitivity_results
# Annotation
adata_st_ecc_dict = {}
adata_st_ecc_dict["CellART"] = annotated_cellart.copy()
adata_st_ecc_dict["10X"] = ad.read_h5ad("10X_scvi_annotated_adata_st_with_ecc.h5ad")
adata_st_ecc_dict["BIDCell"] = ad.read_h5ad("bidcell_scvi_annotated_adata_st_with_ecc.h5ad")
adata_st_ecc_dict["Baysor"] = ad.read_h5ad("baysor_scvi_annotated_adata_st_with_ecc.h5ad")
adata_st_ecc_dict["10X"].obs['celltype'] = adata_st_ecc_dict["10X"].obs["C_scANVI"].replace({"Invasive Tumor": "Malignant", "DCIS 2": "Malignant", "Prolif Invasive Tumor": "Malignant", "DCIS 1": "Malignant", "CD4+ T Cells": "T Cells", "CD8+ T Cells": "T Cells", "Macrophages 1": "Macrophages", "Macrophages 2": "Macrophages", "Myoepi ACTA2+": "Myoepithelial", "Myoepi KRT15+": "Myoepithelial", "IRF7+ DCs": "DCs", "LAMP3+ DCs": "DCs"})
adata_st_ecc_dict["BIDCell"].obs['celltype'] = adata_st_ecc_dict["BIDCell"].obs["C_scANVI"].replace({"Invasive Tumor": "Malignant", "DCIS 2": "Malignant", "Prolif Invasive Tumor": "Malignant", "DCIS 1": "Malignant", "CD4+ T Cells": "T Cells", "CD8+ T Cells": "T Cells", "Macrophages 1": "Macrophages", "Macrophages 2": "Macrophages", "Myoepi ACTA2+": "Myoepithelial", "Myoepi KRT15+": "Myoepithelial", "IRF7+ DCs": "DCs", "LAMP3+ DCs": "DCs"})
adata_st_ecc_dict["Baysor"].obs['celltype'] = adata_st_ecc_dict["Baysor"].obs["C_scANVI"].replace({"Invasive Tumor": "Malignant", "DCIS 2": "Malignant", "Prolif Invasive Tumor": "Malignant", "DCIS 1": "Malignant", "CD4+ T Cells": "T Cells", "CD8+ T Cells": "T Cells", "Macrophages 1": "Macrophages", "Macrophages 2": "Macrophages", "Myoepi ACTA2+": "Myoepithelial", "Myoepi KRT15+": "Myoepithelial", "IRF7+ DCs": "DCs", "LAMP3+ DCs": "DCs"})
for method, adata in adata_st_ecc_dict.items():
# If sparse matrix, convert to dense
if hasattr(adata.X, "todense"):
adata.X = adata.X.toarray()
sensitivity_results_per_method = {}
for method, adata in adata_st_ecc_dict.items():
sensitivity_results = calculate_sensitivity(adata, purified_markers, max_cells_per_type=2000)
sensitivity_results_per_method[method] = sensitivity_results
# Prepare data for sensitivity boxplots
sensitivity_boxplot_data = []
for method, sensitivity_results in sensitivity_results_per_method.items():
for cell_type, sensitivities in sensitivity_results.items():
method_df = pd.DataFrame({"Cell Type": cell_type, "Sensitivity": sensitivities, "Segmentation Method": method})
sensitivity_boxplot_data.append(method_df)
# Concatenate all sensitivity dataframes into one
sensitivity_boxplot_data = pd.concat(sensitivity_boxplot_data)
WARNING: It seems you use rank_genes_groups on the raw count data. Please logarithmize your data before calling rank_genes_groups.
/tmp/ipykernel_3539281/3771815614.py:4: FutureWarning: The behavior of Series.replace (and DataFrame.replace) with CategoricalDtype is deprecated. In a future version, replace will only be used for cases that preserve the categories. To change the categories, use ser.cat.rename_categories instead.
adata_sc.obs['celltype'] = adata_sc.obs['celltype'].replace({"Invasive Tumor": "Malignant", "DCIS 2": "Malignant", "Prolif Invasive Tumor": "Malignant", "DCIS 1": "Malignant", "CD4+ T Cells": "T Cells", "CD8+ T Cells": "T Cells", "Macrophages 1": "Macrophages", "Macrophages 2": "Macrophages", "Myoepi ACTA2+": "Myoepithelial", "Myoepi KRT15+": "Myoepithelial", "IRF7+ DCs": "DCs", "LAMP3+ DCs": "DCs"})
/tmp/ipykernel_3539281/3771815614.py:114: FutureWarning: The behavior of Series.replace (and DataFrame.replace) with CategoricalDtype is deprecated. In a future version, replace will only be used for cases that preserve the categories. To change the categories, use ser.cat.rename_categories instead.
adata_st_ecc_dict["10X"].obs['celltype'] = adata_st_ecc_dict["10X"].obs["C_scANVI"].replace({"Invasive Tumor": "Malignant", "DCIS 2": "Malignant", "Prolif Invasive Tumor": "Malignant", "DCIS 1": "Malignant", "CD4+ T Cells": "T Cells", "CD8+ T Cells": "T Cells", "Macrophages 1": "Macrophages", "Macrophages 2": "Macrophages", "Myoepi ACTA2+": "Myoepithelial", "Myoepi KRT15+": "Myoepithelial", "IRF7+ DCs": "DCs", "LAMP3+ DCs": "DCs"})
/tmp/ipykernel_3539281/3771815614.py:115: FutureWarning: The behavior of Series.replace (and DataFrame.replace) with CategoricalDtype is deprecated. In a future version, replace will only be used for cases that preserve the categories. To change the categories, use ser.cat.rename_categories instead.
adata_st_ecc_dict["BIDCell"].obs['celltype'] = adata_st_ecc_dict["BIDCell"].obs["C_scANVI"].replace({"Invasive Tumor": "Malignant", "DCIS 2": "Malignant", "Prolif Invasive Tumor": "Malignant", "DCIS 1": "Malignant", "CD4+ T Cells": "T Cells", "CD8+ T Cells": "T Cells", "Macrophages 1": "Macrophages", "Macrophages 2": "Macrophages", "Myoepi ACTA2+": "Myoepithelial", "Myoepi KRT15+": "Myoepithelial", "IRF7+ DCs": "DCs", "LAMP3+ DCs": "DCs"})
/tmp/ipykernel_3539281/3771815614.py:116: FutureWarning: The behavior of Series.replace (and DataFrame.replace) with CategoricalDtype is deprecated. In a future version, replace will only be used for cases that preserve the categories. To change the categories, use ser.cat.rename_categories instead.
adata_st_ecc_dict["Baysor"].obs['celltype'] = adata_st_ecc_dict["Baysor"].obs["C_scANVI"].replace({"Invasive Tumor": "Malignant", "DCIS 2": "Malignant", "Prolif Invasive Tumor": "Malignant", "DCIS 1": "Malignant", "CD4+ T Cells": "T Cells", "CD8+ T Cells": "T Cells", "Macrophages 1": "Macrophages", "Macrophages 2": "Macrophages", "Myoepi ACTA2+": "Myoepithelial", "Myoepi KRT15+": "Myoepithelial", "IRF7+ DCs": "DCs", "LAMP3+ DCs": "DCs"})
[29]:
# sensitivity_boxplot_data = pd.read_csv("/import/home2/yhchenmath/Code/Triplet/LOG/XeniumBreastCancer_Annotation_with_feature/epoch_200/sensitivity_boxplot_data.csv")
sensitivity_boxplot_data["Segmentation Method"].value_counts()
[29]:
Segmentation Method
10X 18484
BIDCell 17822
Baysor 17714
CellART 17560
Name: count, dtype: int64
[30]:
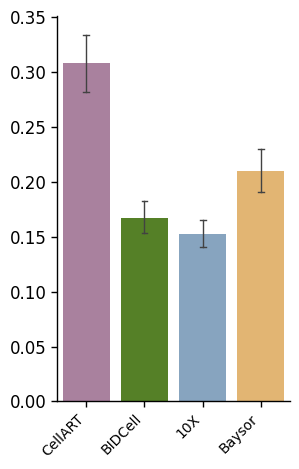
sensitivity_boxplot_data["Segmentation Method"] = pd.Categorical(
sensitivity_boxplot_data["Segmentation Method"],
categories=["CellART", "BIDCell", "10X", "Baysor"]
)
fig, ax = plt.subplots(1, 1, figsize=(3, 5))
plot_bar(sensitivity_boxplot_data, x_col="Segmentation Method", y_col='Sensitivity', hue="Segmentation Method", y_label='', ax=ax, method_colors=method_colors, error_bar=True)
# No x,y labels
ax.set_xlabel('')
ax.set_ylabel('')
# No x tick
# ax.set_xticks([])
# y ticks: 0, 0.2, 0.4, 0.6, 0.8
# ax.set_yticks(np.arange(0.4, 0.62, 0.05))
# Only 1 decimal place
ax.yaxis.set_major_formatter(plt.FuncFormatter(lambda x, _: f'{x:.2f}'))
ax.tick_params(axis='y', labelsize=12)
/tmp/ipykernel_3539281/2228022090.py:39: UserWarning: FixedFormatter should only be used together with FixedLocator
ax.set_xticklabels(ax.get_xticklabels(), rotation=45, ha='right')
[31]:
adata_sc.var.index
[31]:
Index(['IL2RG', 'SNAI1', 'GLIPR1', 'OXTR', 'MYBPC1', 'MUC6', 'PDK4', 'KLRB1',
'RUNX1', 'DSP',
...
'CTH', 'CAV1', 'CD80', 'GATA3', 'ANKRD30A', 'BASP1', 'CD14', 'C5orf46',
'IGF1', 'LEP'],
dtype='object', length=307)
[32]:
ad_st_dict = {
"10X_tangram": annotated_10X_tangram.copy(),
"10X_scvi": annotated_10X_scvi.copy(),
"CellART": annotated_cellart.copy()
}
# Varnames: Upper case
for method in ad_st_dict.keys():
ad_st_dict[method].var.index = ad_st_dict[method].var.index.str.upper()
# Only varnames in the gene map are used
adata_sc = adata_sc[:, annotated_cellart.var_names].copy()
adata_sc.var.index = adata_sc.var.index.str.upper()
all_avg_df = {}
for method in ad_st_dict.keys():
avg_df = pd.DataFrame(index=np.unique(ad_st_dict[method].obs["celltype"]), columns=ad_st_dict[method].var.index)
ad_temp= ad_st_dict[method].copy()
# Normalize total
sc.pp.normalize_total(ad_temp, target_sum=1e4)
# Log1p
sc.pp.log1p(ad_temp)
for cell_type in np.unique(ad_temp.obs["celltype"]):
ad = ad_temp[ad_temp.obs["celltype"] == cell_type]
avg_df.loc[cell_type] = ad.X.mean(axis=0)
all_avg_df[method] = avg_df
avg_sc = pd.DataFrame(index=np.unique(adata_sc.obs["celltype"]), columns = adata_sc.var.index)
ad_temp = adata_sc.copy()
# Scale
sc.pp.normalize_total(ad_temp, target_sum=1e4)
# Log1p
sc.pp.log1p(ad_temp)
for cell_type in np.unique(ad_temp.obs["celltype"]):
ad = ad_temp[ad_temp.obs["celltype"] == cell_type]
avg_sc.loc[cell_type] = ad.X.mean(axis=0)
# Same column order
for method in all_avg_df.keys():
all_avg_df[method] = all_avg_df[method][avg_sc.columns]
# Pearson correlation for each method and each Celltype
from scipy.stats import pearsonr
all_corr_df = {}
for method in all_avg_df.keys():
corr_df = pd.DataFrame(index=np.unique(adata_sc.obs["celltype"]), columns=all_avg_df[method].index)
for cell_type in np.unique(adata_sc.obs["celltype"]):
for method_cell_type in all_avg_df[method].index:
corr_df.loc[cell_type, method_cell_type] = pearsonr(all_avg_df[method].loc[method_cell_type], avg_sc.loc[cell_type])[0]
all_corr_df[method] = corr_df
# Convert to float
all_corr_df[method] = all_corr_df[method].astype(float)
WARNING: adata.X seems to be already log-transformed.
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/scanpy/preprocessing/_normalization.py:234: UserWarning: Some cells have zero counts
warn(UserWarning("Some cells have zero counts"))
[33]:
celltype_corr_df = pd.DataFrame(index=list(all_corr_df.keys()), columns=np.unique(adata_sc.obs["celltype"]))
for cell_type in np.unique(adata_sc.obs["celltype"]):
for method in all_corr_df.keys():
celltype_corr_df.loc[method, cell_type] = all_corr_df[method].loc[cell_type, cell_type]
[34]:
# Calculate the average pearson correlation for each method
corr_avg = {}
for method in all_corr_df.keys():
temp = []
for cell_type in all_corr_df[method].index:
temp.append(all_corr_df[method].loc[cell_type, cell_type])
corr_avg[method] = np.mean(temp)
[35]:
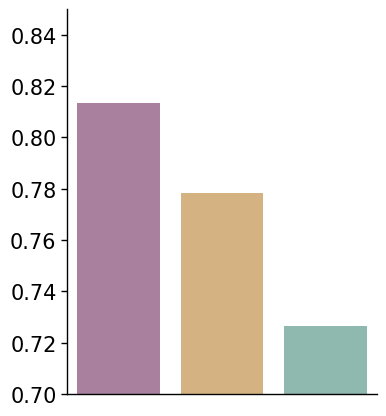
color_dict = {
"CellART": "#B07AA1",
"10X_tangram": "#E2B573",
"10X_scvi": "#88BFB1",
}
df = {
"Method": ["CellART", "10X_tangram", "10X_scvi"],
"Value": [corr_avg["CellART"], corr_avg["10X_tangram"], corr_avg["10X_scvi"]]
}
fig, ax = plt.subplots(1, 1, figsize=(4, 5))
plot_bar(df, x_col="Method", y_col='Value', hue="Method", y_label='', ax=ax, method_colors=color_dict, error_bar=False)
# No x,y labels
ax.set_xlabel('')
ax.set_ylabel('')
ax.set_ylim(0.7, 0.85)
# No x tick
ax.set_xticks([])
# y ticks: 0, 0.2, 0.4, 0.6, 0.8
# ax.set_yticks(np.arange(0.4, 0.62, 0.05))
# Only 1 decimal place
ax.yaxis.set_major_formatter(plt.FuncFormatter(lambda x, _: f'{x:.2f}'))
ax.tick_params(axis='y', labelsize=15)
/tmp/ipykernel_3539281/2228022090.py:39: UserWarning: FixedFormatter should only be used together with FixedLocator
ax.set_xticklabels(ax.get_xticklabels(), rotation=45, ha='right')
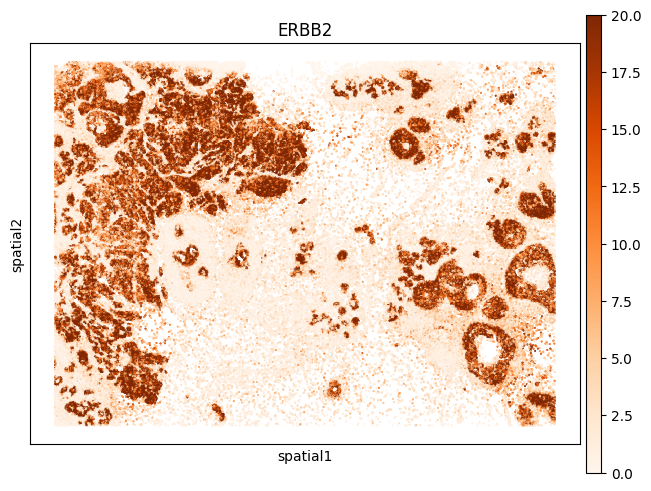
Spatial gene
[36]:
# Spatial gene
import squidpy as sq
annotated_cellart.obsm["spatial"] = np.array(annotated_cellart.obs[['center_y', 'center_x']])
sq.gr.spatial_neighbors(annotated_cellart)
sq.gr.spatial_autocorr(annotated_cellart, mode="moran")
sq.pl.spatial_scatter(
annotated_cellart,
shape=None,
color=["ERBB2"],
size=0.5,
cmap = "Oranges",
vmax = 20,
dpi=100
)
WARNING: Please specify a valid `library_id` or set it permanently in `adata.uns['spatial']`
[37]:
annotated_10X_scvi.X = annotated_10X_scvi.layers["counts"]
annotated_10X_scvi.obsm["spatial"] = np.array(annotated_10X_scvi.obs[['center_y', 'center_x']])
sq.gr.spatial_neighbors(annotated_10X_scvi)
sq.gr.spatial_autocorr(annotated_10X_scvi, mode="moran")
sq.pl.spatial_scatter(
annotated_10X_scvi,
shape=None,
color=["ERBB2"],
size=0.5,
cmap = "Oranges",
vmax = 10,
dpi=100
)
WARNING: Please specify a valid `library_id` or set it permanently in `adata.uns['spatial']`

Velocity
[38]:
annotated_cellart_subtype = sc.read_h5ad("cellart_subtype_cell_deconv.h5ad")
cellart_mask_subtype = np.load("cellart_subtype_segmentation_mask.npy").astype("int32")
cellart_nuclei = np.load("cellart_subtype_nuclei.npy") # U
append_xenium_boundary(cellart_mask_subtype, sdata, "cellart_boundary_subtype", celltype = annotated_cellart_subtype.obs["celltype"])
append_xenium_boundary(cellart_nuclei, sdata, "cellart_nuclei_boundary")
/tmp/ipykernel_3539281/2510591147.py:92: UserWarning: Geometry is in a geographic CRS. Results from 'centroid' are likely incorrect. Use 'GeoSeries.to_crs()' to re-project geometries to a projected CRS before this operation.
center_df = sdata.shapes[append_name].centroid
[39]:
seg_transformation = sd.transformations.get_transformation(sdata.shapes["nucleus_boundaries"])
translation = sd.transformations.Translation([6, 6.5], axes=("x", "y"))
sequence = sd.transformations.Sequence([translation, seg_transformation])
sd.transformations.set_transformation(sdata.shapes["cellart_boundary_subtype"], sequence, to_coordinate_system="global")
sd.transformations.set_transformation(sdata.shapes["cellart_nuclei_boundary"], sequence, to_coordinate_system="global")
[40]:
x_min, x_max, y_min, y_max = 15600, 15950, 32325, 32675
sdata_roi_2 = sdata.query.bounding_box(
min_coordinate=[x_min, y_min], max_coordinate=[x_max, y_max], axes=("y", "x"), target_coordinate_system="global"
)
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/anndata/_core/anndata.py:183: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
[41]:
k = 'cellart_boundary_subtype'
# sdata_roi_2.shapes[k].celltype = sdata_roi_2.shapes[k].celltype.map(mapping)
# Drop nan
sdata_roi_2.shapes[k].dropna(subset=["celltype"], inplace=True)
ct_col = sdata_roi_2.shapes[k].celltype
cts = sdata_roi_2.shapes[k].celltype.unique()
for ct in cts:
sdata_roi_2.shapes[f"{k}_{ct.replace(' ', '_').replace('(', '').replace(')', '')}"] = \
sdata_roi_2.shapes[k][ct_col == ct]
/tmp/ipykernel_3539281/1554620027.py:4: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame
See the caveats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#returning-a-view-versus-a-copy
sdata_roi_2.shapes[k].dropna(subset=["celltype"], inplace=True)
[42]:
# Plot cellart
celltype_mapping = {
"Unassigned": '#D3D3D3',
'Invasive Tumor': '#E477C1',
'Prolif Invasive Tumor': '#289E68',
'DCIS 2': '#FF7F0D',
'DCIS 1': '#AA41FC',
}
def plot_annotation(sdata, shape_key, ax, title):
tmp = sdata.pl.render_images("he_image")
draw_cts = sdata.shapes[shape_key].celltype.unique().tolist()
for ct in draw_cts:
color = celltype_mapping[ct]
tmp = tmp.pl.render_shapes(
f"{shape_key}_{ct.replace(' ', '_').replace('(', '').replace(')', '')}",
color=color, fill_alpha=0.45, outline_width=3, outline_alpha=1, outline_color = color
)
tmp = tmp.pl.render_shapes(
f"cellart_nuclei_boundary",
color="w", fill_alpha=0.0, outline_width=3, outline_alpha=1, outline_color = "w"
)
tmp.pl.show(coordinate_systems="global", ax=ax, title="", frameon=False, legend_loc='none')
ax.axis('off')
[43]:
# plt.style.use('default')
import matplotlib.pyplot as plt
fig, ax = plt.subplots(1, 1, figsize=(6,6))
plot_annotation(sdata_roi_2, k, ax, "")
ax.set_xlim(y_min, y_max)
ax.set_ylim(x_min, x_max)
# plt.tight_layout()
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Value for parameter 'color' appears to be a color, using it as such.
Clipping input data to the valid range for imshow with RGB data ([0..1] for floats or [0..255] for integers).
[43]:
(15600.0, 15950.0)
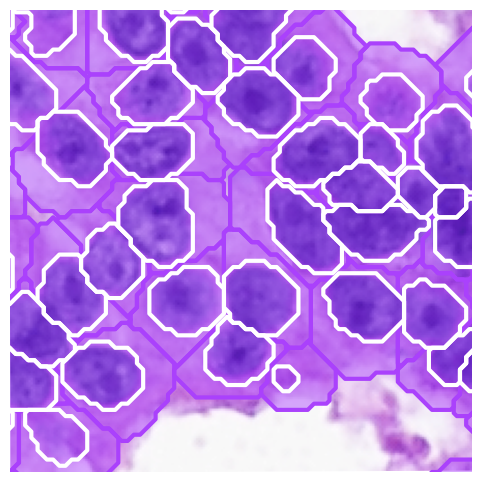
[44]:
import geopandas as gpd
gdf_cell = sdata.shapes["cellart_boundary_subtype"]
gdf_cell["x"] = sdata.points["cellart_boundary_subtype_centroid"].compute()['x'].values
gdf_cell["y"] = sdata.points["cellart_boundary_subtype_centroid"].compute()['y'].values
gdf_nuclei = sdata.shapes["cellart_nuclei_boundary"]
common_ids = gdf_cell.index.intersection(gdf_nuclei.index)
cells_common = gdf_cell.loc[common_ids]
nuclei_common = gdf_nuclei.loc[common_ids]
cytoplasm_geometry = cells_common.geometry.difference(nuclei_common.geometry)
[45]:
# As a geodataframe
gdf_cyto = gpd.GeoDataFrame({
'cell_id': cells_common.index,
'geometry': cytoplasm_geometry
})
gdf_cyto = gdf_cyto.set_index("cell_id")
[ ]:
from spatialdata.models import ShapesModel
sdata["cyto"] = ShapesModel.parse(gdf_cyto)
# Set transformation
sd.transformations.set_transformation(sdata.shapes["cyto"], sequence, to_coordinate_system="global")
sdata["nuclei_common"] = ShapesModel.parse(nuclei_common)
sd.transformations.set_transformation(sdata.shapes["nuclei_common"], sequence, to_coordinate_system="global")
[ ]:
import sopa
adata_nuclei = sopa.aggregation.count_transcripts(sdata, points_key='transcripts', shapes_key="nuclei_common")
adata_cyto = sopa.aggregation.count_transcripts(sdata, points_key='transcripts', shapes_key="cyto")
# adata_nuclei.write_h5ad("/import/home2/yhchenmath/Code/Triplet/LOG/XeniumBreastCancer_Annotation_with_feature/nuclei.h5ad")
adata_cyto.obs["celltype"] = cells_common.celltype.values
adata_cyto.obs["x"] = cells_common.x.values
adata_cyto.obs["y"] = cells_common.y.values
# # Save
# adata_cyto.write_h5ad("/import/home2/yhchenmath/Code/Triplet/LOG/XeniumBreastCancer_Annotation_with_feature/cyto.h5ad")
# import scanpy as sc
# adata_cyto = sc.read_h5ad("/import/home2/yhchenmath/Code/Triplet/LOG/XeniumBreastCancer_Annotation_with_feature/cyto.h5ad")
# adata_nuclei = sc.read_h5ad("/import/home2/yhchenmath/Code/Triplet/LOG/XeniumBreastCancer_Annotation_with_feature/nuclei.h5ad")
[46]:
# Plot svt
celltype_mapping = {
"Unassigned": '#D3D3D3',
'Invasive Tumor': '#E477C1',
'Prolif Invasive Tumor': '#289E68',
'DCIS 2': '#FF7F0D',
'DCIS 1': '#AA41FC',
}
[47]:
adata = sc.read_h5ad("cellart_subtype_cell_deconv.h5ad")
# adata = adata[adata.obs["celltype"].isin(["DCIS 1", "DCIS 2", "Invasive Tumor", "Prolif Invasive Tumor"]), :].copy()
mapping = {
"Invasive Tumor": "Invasive Tumor",
"Prolif Invasive Tumor": "Prolif Invasive Tumor",
"DCIS 2": "DCIS 2",
"DCIS 1": "DCIS 1",
}
for celltype in adata.obs["celltype"].unique():
if celltype not in mapping:
mapping[celltype]="Unassigned"
adata.obs["celltype"] = adata.obs["celltype"].map(mapping)
# Filtered
sc.pp.filter_cells(adata, min_counts=10)
[48]:
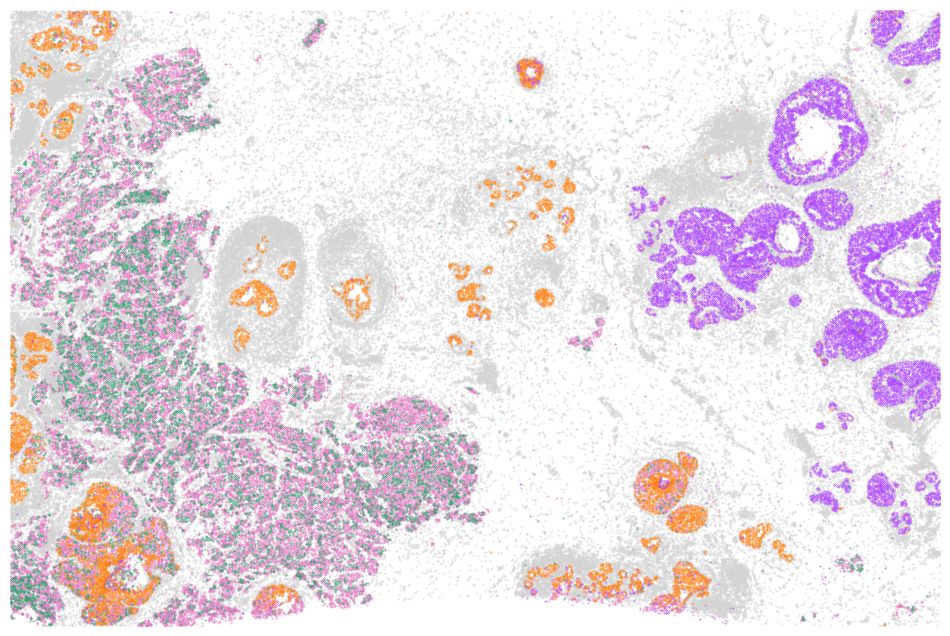
from matplotlib.gridspec import GridSpec
from matplotlib.lines import Line2D
import matplotlib.pyplot as plt
df = adata.obs
fig, ax1 = plt.subplots(1, 1, figsize=(12,8))
from matplotlib.patches import Rectangle
celltype_names = list(celltype_mapping.keys())
# selected_celltype = ["Tumor II", "Tumor III", "Tumor V"]
selected_celltype = celltype_names
for i in range(len(celltype_names)):
# (0,0) is on the top left corner
if celltype_names[i] not in selected_celltype:
continue
sub_df = df[df["celltype"] == celltype_names[i]]
ax1.scatter(sub_df["y"], sub_df["x"], s=0.1, label=celltype_names[i], color=celltype_mapping[celltype_names[i]])
# ax1.invert_yaxis()
ax1.axis("off")
ax1.set_xlim(df["y"].min(), df["y"].max())
ax1.set_ylim(df["x"].max(), df["x"].min())
ax1.invert_yaxis()
plt.show()
[49]:
adata_cyto = sc.read_h5ad("cyto.h5ad")
adata_nuclei = sc.read_h5ad("nuclei.h5ad")
adata = adata_cyto.copy()
adata.layers["unspliced"] = adata_nuclei.X
adata.layers["spliced"] = adata_cyto.X
# Filtered cells umi < 50
sc.pp.filter_cells(adata, min_counts=100)
adata = adata[adata.obs["celltype"].isin(["DCIS 1", "DCIS 2", "Invasive Tumor", "Prolif Invasive Tumor"]), :].copy()
import scvelo as scv
scv.pp.filter_and_normalize(adata, min_shared_counts=20)
# Remove duplicated cell
scv.pp.remove_duplicate_cells(adata)
scv.pp.moments(adata)
scv.tl.velocity(adata, mode='stochastic')
scv.tl.velocity_graph(adata)
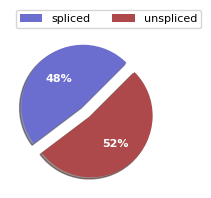
scv.pl.proportions(adata)
# UMAP
sc.pp.neighbors(adata, use_rep="X_pca")
sc.tl.umap(adata)
Filtered out 29 genes that are detected 20 counts (shared).
Normalized count data: X, spliced, unspliced.
Logarithmized X.
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/scvelo/preprocessing/utils.py:705: DeprecationWarning: `log1p` is deprecated since scVelo v0.3.0 and will be removed in a future version. Please use `log1p` from `scanpy.pp` instead.
log1p(adata)
/tmp/ipykernel_3539281/2510900289.py:16: DeprecationWarning: Automatic neighbor calculation is deprecated since scvelo==0.4.0 and will be removed in a future version of scVelo. Please compute neighbors first with Scanpy.
scv.pp.moments(adata)
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/scvelo/preprocessing/moments.py:71: DeprecationWarning: `neighbors` is deprecated since scvelo==0.4.0 and will be removed in a future version of scVelo. Please compute neighbors with Scanpy.
neighbors(
computing neighbors
2025-12-13 20:08:16.556780: I tensorflow/core/platform/cpu_feature_guard.cc:182] This TensorFlow binary is optimized to use available CPU instructions in performance-critical operations.
To enable the following instructions: AVX2 AVX512F FMA, in other operations, rebuild TensorFlow with the appropriate compiler flags.
2025-12-13 20:08:18.555037: W tensorflow/compiler/tf2tensorrt/utils/py_utils.cc:38] TF-TRT Warning: Could not find TensorRT
finished (0:00:55) --> added
'distances' and 'connectivities', weighted adjacency matrices (adata.obsp)
computing moments based on connectivities
finished (0:00:01) --> added
'Ms' and 'Mu', moments of un/spliced abundances (adata.layers)
computing velocities
finished (0:00:02) --> added
'velocity', velocity vectors for each individual cell (adata.layers)
computing velocity graph (using 1/44 cores)
finished (0:00:55) --> added
'velocity_graph', sparse matrix with cosine correlations (adata.uns)
[50]:
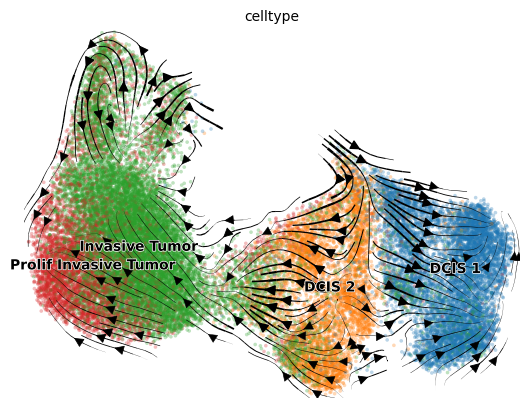
scv.pl.velocity_embedding_stream(adata, basis='umap', color='celltype', dpi=100, arrow_size=1.5, legend_fontsize=10)
computing velocity embedding
finished (0:00:08) --> added
'velocity_umap', embedded velocity vectors (adata.obsm)
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/scvelo/plotting/utils.py:68: DeprecationWarning: is_categorical_dtype is deprecated and will be removed in a future version. Use isinstance(dtype, pd.CategoricalDtype) instead
return isinstance(c, str) and c in data.obs.keys() and cat(data.obs[c])
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/scvelo/plotting/utils.py:68: DeprecationWarning: is_categorical_dtype is deprecated and will be removed in a future version. Use isinstance(dtype, pd.CategoricalDtype) instead
return isinstance(c, str) and c in data.obs.keys() and cat(data.obs[c])