Reproduce results in Figure 6
NOTE: Same datasets in Figure 5. Visualizations of figure 6b and c are in figure 5 notebook with section named
Visualization for figure 6b,c.
Related dataset:
Reference of CRC can be downloaded here
VisiumHD colorectal cancer are training with epoch settings: epochs = 400 seg_training_epochs = 15 deconv_warmup_epochs = 200 and Xenium coloretal cancer are training with epochs settings: epochs = 200 seg_training_epochs = 10 deconv_warmup_epochs = 100
All the data for reproducing the result can be downloaded from Zenodo. The data used in this notebook is under the folder figure_6.
[38]:
import os
# Download the data from the link above to this folder and unzip it, can be changed to your own path
os.chdir("/import/home3/yhchenmath/Dataset/CellARTPaper/figure_6/")
Characterization of TAMs
[39]:
import pandas as pd
from scipy.sparse import coo_matrix
import squidpy as sq
import matplotlib.pyplot as plt
import numpy as np
import scanpy as sc
[40]:
adata_svt = sc.read_h5ad("cellart_crc_adata.h5ad")
adata_svt.obsm["spatial"] = adata_svt.obs[["x", "y"]].values
[41]:
import pandas as pd
from scipy.sparse import coo_matrix
import squidpy as sq
adata = adata_svt.copy()
sq.gr.spatial_neighbors(adata, n_neighs = 100)
sparse_matrix = adata.obsp["spatial_distances"].tocoo()
# Create DataFrame from COO matrix
df_dist = pd.DataFrame({
"value": sparse_matrix.data,
"x": sparse_matrix.col,
"y": sparse_matrix.row
})
df_dist.columns = ["distance", "cell_1_index", "cell_2_index"]
distance_threshold = 100
celltypes = adata.obs["celltype"]
df_dist["cell_1_type"] = celltypes.iloc[df_dist["cell_1_index"]].values
df_dist["cell_2_type"] = celltypes.iloc[df_dist["cell_2_index"]].values
df_close = df_dist[df_dist["distance"] < distance_threshold]
mac_near_tumor = df_close[
((df_close["cell_1_type"] == "Macrophage") & (df_close["cell_2_type"].str.startswith("Tumor III"))) |
((df_close["cell_2_type"] == "Macrophage") & (df_close["cell_1_type"].str.startswith("Tumor III")))
]
cd4_t_near_tumor = df_close[
((df_close["cell_1_type"] == "CD4 T cell") & (df_close["cell_2_type"].str.startswith("Tumor III"))) |
((df_close["cell_2_type"] == "CD4 T cell") & (df_close["cell_1_type"].str.startswith("Tumor III")))
]
cd8_t_near_tumor = df_close[
((df_close["cell_1_type"] == "CD8 Cytotoxic T cell") & (df_close["cell_2_type"].str.startswith("Tumor III"))) |
((df_close["cell_2_type"] == "CD8 Cytotoxic T cell") & (df_close["cell_1_type"].str.startswith("Tumor III")))
]
# All index
mac_tumor_indices = pd.concat([
mac_near_tumor["cell_1_index"],
mac_near_tumor["cell_2_index"]
]).drop_duplicates().sort_values()
cd4_tumor_indices = pd.concat([
cd4_t_near_tumor["cell_1_index"],
cd4_t_near_tumor["cell_2_index"]
]).drop_duplicates().sort_values()
cd8_tumor_indices = pd.concat([
cd8_t_near_tumor["cell_1_index"],
cd8_t_near_tumor["cell_2_index"]
]).drop_duplicates().sort_values()
mac_tumor_cell_names = adata.obs_names[mac_tumor_indices]
# cd4_tumor_cell_names = adata.obs_names[cd4_tumor_indices]
# cd8_tumor_cell_names = adata.obs_names[cd8_tumor_indices]
print(len(mac_tumor_cell_names))
# Only select macrophage and tumor cells
adata_boundary = adata[mac_tumor_cell_names].copy()
# adata_boundary.write_h5ad("/import/home2/yhchenmath/Code/SVTBenchmarking/figure_refined/adata_lr.h5ad")
94616
[42]:
# Select normal macrophage
adata_normal = adata[adata.obs["celltype"] == "Macrophage"].copy()
# Not in mac_tumor_cell_names
normal_cell_names = adata_normal.obs_names[~adata_normal.obs_names.isin(mac_tumor_cell_names)]
adata_normal = adata_normal[normal_cell_names].copy()
# Select all the tumor cells
tumor_cell_names = adata.obs_names[adata.obs["celltype"].str.startswith("Tumor III")]
adata_tumor = adata[tumor_cell_names].copy()
[46]:
import anndata
adata_tam = adata_boundary[adata_boundary.obs["celltype"] == "Macrophage"].copy()
adata_tam.obs["celltype"] = "TAM"
adata_normal.obs["celltype"] = "Normal Macrophage"
adata_tumor.obs["celltype"] = "Tumor"
adata_plot = anndata.concat([adata_tam, adata_normal, adata_tumor])
adata_others = adata[~adata.obs_names.isin(adata_plot.obs_names)].copy()
adata_whole = anndata.concat([adata_plot, adata_others])
# Save adata_whole
# adata_whole.write_h5ad("/import/home2/yhchenmath/Code/SVTPaper/figure_5/lr/adata_whole.h5ad")
[47]:
# Plot
from matplotlib.gridspec import GridSpec
from matplotlib.lines import Line2D
import matplotlib.pyplot as plt
def plot_specific_celltype_gene(adata, mapping_dict, x_col = "x", y_col = "y", s = 3, revert_y = False, revert_x = False, celltype_col = "celltype"):
fig, ax = plt.subplots(1, 4, figsize=(16, 8))
for i in range(4):
ax[i].set_axis_off()
gs = GridSpec(1, 4, figure=fig)
ax1 = fig.add_subplot(gs[0, :3])
ax2 = fig.add_subplot(gs[0, 3])
# fig, ax1 = plt.subplots(1, 1, figsize=(12, 8))
plot_dict = {}
# Mapping dict: "[ct] [gene]"
for k in mapping_dict:
ct, gene = k.split(" ")
ct = ct.replace("_", " ")
# Select those cells: celltye is ct and gene expression of gene is not zero
temp_adata = adata[(adata.obs[celltype_col] == ct)]
if gene != "all":
temp_adata = temp_adata[temp_adata[:, gene].X > 0]
else:
pass
plot_dict[k] = temp_adata.copy()
ax1.scatter(adata.obs[y_col], adata.obs[x_col], s=s / 2, color="lightgray")
for k in plot_dict:
temp_adata = plot_dict[k]
ax1.scatter(temp_adata.obs[y_col], temp_adata.obs[x_col], s=s, alpha=0.4, edgecolor=mapping_dict[k], lw=0.8, label=k, color =mapping_dict[k])
ax1.scatter(temp_adata.obs[y_col], temp_adata.obs[x_col], s=s, alpha=1, edgecolor=mapping_dict[k], lw=0.8, label=k, c="none")
ax1.axis("off")
ax1.set_xlim(adata.obs[x_col].min(), adata.obs[x_col].max())
ax1.set_ylim(adata.obs[y_col].min(), adata.obs[y_col].max())
if revert_x:
ax1.invert_xaxis()
if revert_y:
ax1.invert_yaxis()
# Add legend elements (example)
legend_elements = [
Line2D(
[0], [0],
marker='o',
linestyle='None',
color='w',
label=label.replace("_", " ").replace(" all", ""),
markerfacecolor=color,
markeredgecolor='k',
markersize=8
) for label, color in mapping_dict.items()
]
# Add the legend below the entire figure, centered horizontally with [0, 1] subfigures
ax2.legend(
handles=legend_elements,
loc='center', # Center the legend within the bounding box
bbox_to_anchor=(0.48, 0.45), # Center of ax2 (0.5, 0.5 is the middle of the axis)
ncol=2, # Number of columns for the legend
handletextpad=0.35, # Spacing between marker and text
columnspacing=1, # Spacing between legend columns
prop={'size': 12, 'style': 'italic'}, # Font size and style
frameon=False # No border for the legend
)
ax2.axis("off") # Hide the axis for ax2
# Adjust layout to prevent overlap
plt.tight_layout(rect=[0, 0.15, 1, 1]) # Leave space for the legend below the plots
plt.show()
return plot_dict
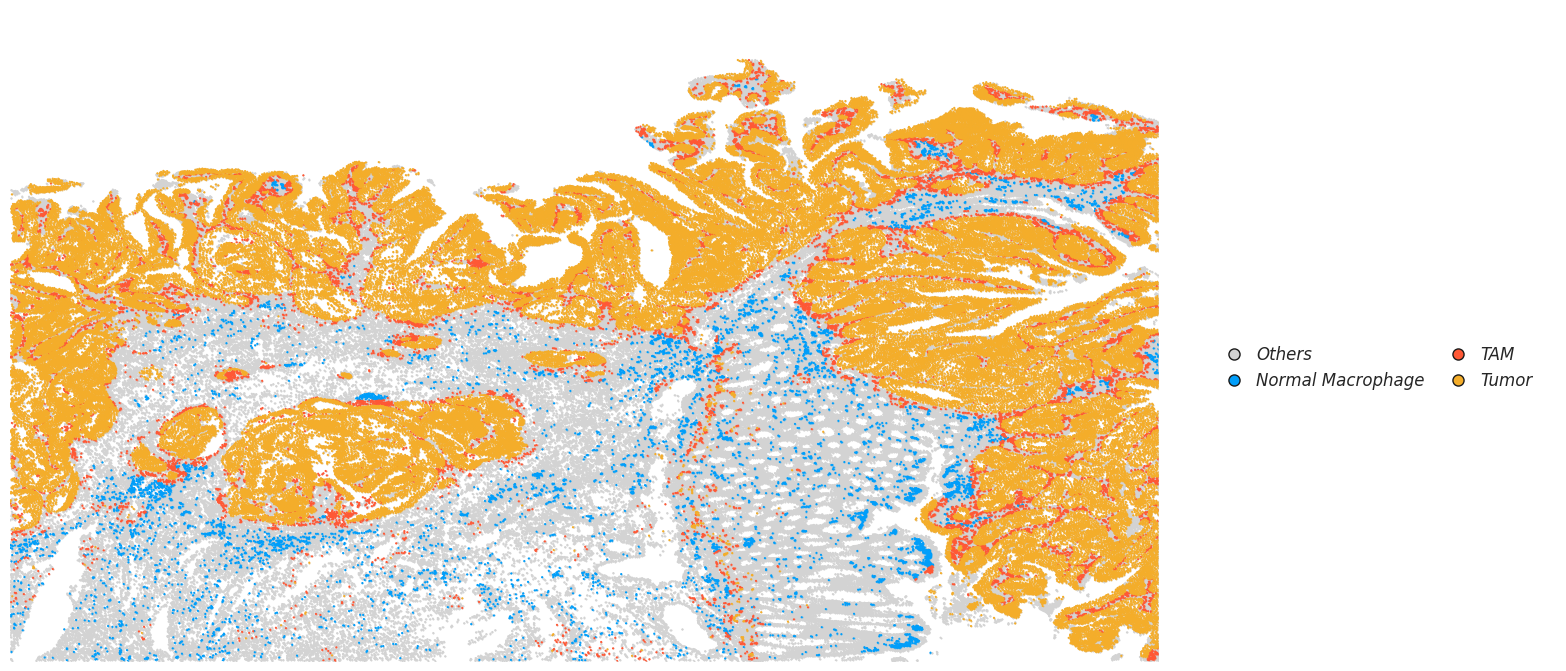
[48]:
mapping_dict = {
"Others all": "lightgray",
"Normal_Macrophage all": "#009EFA",
"TAM all": "#FF5835",
"Tumor all": "#F4AD2A",
}
adata_others_temp = adata_others.copy()
adata_others_temp.obs["celltype"] = "Others"
adata_combined = anndata.concat([adata_plot, adata_others_temp])
_ = plot_specific_celltype_gene(adata_combined, mapping_dict, x_col = "x", y_col = "y", s = 0.5, revert_y = False, revert_x= False)
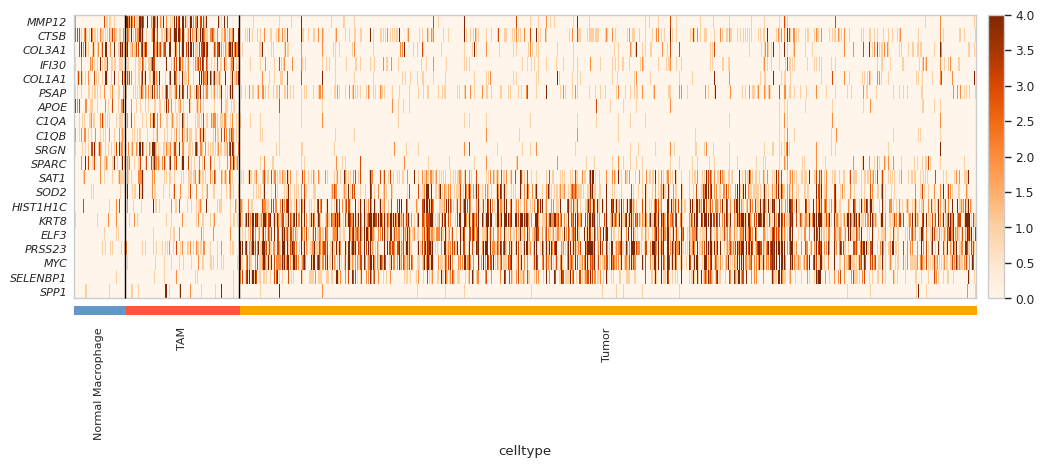
[49]:
selected_gene = [
"MMP12","CTSB", "COL3A1", "IFI30", "COL1A1", "PSAP", "APOE", "C1QA", "C1QB", "SRGN", "SPARC"
,"SAT1", "SOD2", "HIST1H1C", "KRT8", "ELF3", "PRSS23", "MYC", "SELENBP1", "SPP1"
]
gene_list = selected_gene
# As type categorical
adata_plot.obs['celltype'] = adata_plot.obs['celltype'].astype('category')
adata_plot.obs['celltype'] = adata_plot.obs['celltype'].cat.reorder_categories(
["Normal Macrophage", "TAM", "Tumor"],
ordered=True # optional
)
adata_plot.uns["celltype_colors"] = ["#6297CA", "#FF5733", "#FFA500"]
# Only show invasive tumor and Prolif Invasive Tumor
hm = sc.pl.heatmap(adata_plot, gene_list, groupby='celltype', cmap='Oranges', show_gene_labels=True, vmax=4, swap_axes=True, show=False, figsize=(12,4))
ax = hm['heatmap_ax']
for l in ax.get_yticklabels():
l.set_style("italic")
# Show with high dpi
plt.show(block=True)
[50]:
import scanpy as sc
from collections.abc import Iterable
from typing import Union, List
import itertools
from mpl_toolkits.axes_grid1 import make_axes_locatable
from matplotlib import pyplot as plt
import math
from pathlib import Path
# from sg_utils.pl.utils import lighten_color
def format_ax(
fig, ax,
style="umap",
title="",
cbar=True,
dim_label="UMAP",
fs=12,
lw=1.5,
arrow_len=0.2,
draw_arrows=True,
):
ax.set_facecolor('white')
ax.set_xticklabels([])
ax.set_yticklabels([])
ax.get_xaxis().set_visible(False)
ax.get_yaxis().set_visible(False)
ax.grid(False)
ax.spines[list(ax.spines)].set_visible(False)
if style == "umap":
change_aspect(ax)
if draw_arrows:
arrowed_spines(ax, arrow_len, text=dim_label, fs=fs, lw=lw)
ax.set_title(title, weight="bold")
if cbar:
format_cbar(fig, ax)
def format_cbar(fig, ax):
cbar = ax.get_children()[0].colorbar
if cbar:
cbar.remove()
data = ax.get_children()[0]
# Create colorbar ax
bbox = ax.get_position()
cax = fig.add_axes([
bbox.x1+bbox.width*0.025, #min x
bbox.y0+bbox.height*0.25, #min y
bbox.width*0.03, #width
bbox.height*0.5 #height
])
cax.grid(False)
new_cbar = fig.colorbar(
data, ax=ax, cax=cax,
)
new_cbar.outline.set_visible(False)
if not cbar:
bbox = ax.get_position()
ax.get_children()[0].colorbar.remove()
ax.set_position(bbox)
def change_aspect(ax):
# Reset x and y limits for square plotting
xmin, xmax = ax.get_xlim()
xrange = xmax - xmin
xcenter = (xrange/2) + xmin
ymin, ymax = ax.get_ylim()
yrange = ymax - ymin
ycenter = (yrange/2) + ymin
axrange = max(xrange, yrange)/2
xmin = xcenter - (axrange)
xmax = xcenter + (axrange)
ax.set_xlim(xmin, xmax)
ymin = ycenter - (axrange)
ymax = ycenter + (axrange)
ax.set_ylim(ymin, ymax)
ax.set_aspect('equal', adjustable = 'box')
def arrowed_spines(
ax,
length = 0.2,
text = None,
fs = None,
lw = 1.5,
):
xmin, xmax = ax.get_xlim()
ymin, ymax = ax.get_ylim()
hw = 1./30.*(ymax-ymin)
hl = 1./30.*(xmax-xmin)
lw = lw # axis line width
ohg = 0.0 # arrow overhang
ax.spines[list(ax.spines)].set_visible(False)
ax.arrow(
xmin, ymin, (xmax-xmin)*length, 0, fc='k', ec='k', lw = lw,
head_width=hw, head_length=hl, overhang = ohg,
length_includes_head= True, clip_on = False
)
ax.arrow(
xmin, ymin, 0, (ymax-ymin)*length, fc='k', ec='k', lw = lw,
head_width=hw, head_length=hl, overhang = ohg,
length_includes_head= True, clip_on = False
)
if fs == None:
fs = plt.rcParams["xtick.labelsize"]
ax.text(
s=f"{text}1",
y=ymin-(ymax-ymin)*0.05, x=xmin+(xmax-xmin)*length/2,
ha="center", va="top",
fontsize = fs
)
ax.text(
s=f"{text}2",
x=xmin-(xmax-xmin)*0.05, y=ymin+(ymax-ymin)*length/2,
ha="right", va="center", rotation=90,
fontsize = fs
)
ax.set_xlim(xmin, xmax)
ax.set_ylim(ymin, ymax)
def plot_embedding(
adata: sc.AnnData,
features: Union[str, List[str]],
basis: str = 'X_umap',
palette: str = "tab20",
cmap: str = "plasma",
titles: Union[str, List[str]] = None,
ncols: int = 5,
dim: int = 5,
layer: str = "imputed",
dim_label = "UMAP",
ax = None,
fs: int = 12,
lw: float = 1.5,
arrow_len: float = 0.2,
draw_arrows=False,
rasterized=False,
cbar=True,
**kwargs,
):
iterify = lambda x: x if isinstance(x, Iterable) and not isinstance(x, str) else [x]
features = iterify(features)
titles = iterify(titles)
if not ax:
nrows = math.ceil(len(features))
fig, axes = plt.subplots(
nrows, ncols,
figsize=(dim*ncols,dim*nrows),
)
fig.tight_layout(pad=dim*0.75)
axes = axes.flat if isinstance(axes, Iterable) else [axes]
else:
assert (len(features)==1) and (len(titles)==1)
fig = ax.get_figure()
axes = [ax]
for ax, feature, title in itertools.zip_longest(axes, features, titles):
if not title: title = feature
if feature:
sc.pl.embedding(
adata,
basis=basis,
color=feature,
ax=ax,
show=False,
palette=palette, cmap=cmap,
layer=layer,
colorbar_loc=None,
**kwargs,
)
if rasterized:
ax.get_children()[0].set_rasterized(True)
format_ax(
fig, ax, style="umap",
title=title, dim_label=dim_label, fs=fs,
arrow_len=arrow_len, lw=lw, draw_arrows=draw_arrows,
)
else:
ax.set_visible(False)
return fig
def saturate(c, s=1.0):
from matplotlib import colors
from collections.abc import Iterable
# Assumed to be hex if string
if isinstance(c, str):
rgb = colors.to_rgb(c)
hex = True
# Assumed to be RGB if iterable
elif isinstance(c, Iterable):
rgb = c
hex = False
hsv = colors.rgb_to_hsv(rgb)
hsv[1] *= s
c = colors.hsv_to_rgb(hsv)
if hex:
return colors.to_hex(c)
else:
return c
[26]:
ad_st = adata_plot.copy()
# Filtered min counts
sc.pp.filter_genes(ad_st, min_counts=20)
sc.pp.pca(ad_st)
sc.pp.neighbors(ad_st)
sc.tl.umap(ad_st, min_dist=0.3)
celltype_mapping = {
"Normal Macrophage": "#009EFA",
"TAM": "#FF5835",
"Tumor": "#F4AD2A",
}
fig, ax = plt.subplots(1, 1, figsize = (3,3))
_ = plot_embedding(
ad_st,
features='celltype',
basis="X_umap",
palette=celltype_mapping,
ax=ax,
dim_label="UMAP",
fs=12,
lw=1.5,
arrow_len=0.2,
draw_arrows=True,
# Other
legend_loc = "none",
size = 3
)
[51]:
import matplotlib.pyplot as plt
import pandas as pd
import seaborn as sns
sns.set_style("whitegrid")
# selected_gene = "COL3A1" # Tumor+, Boundary +
# selected_gene = "PSAP" # Boundary +, HD Only
# Set color mapping
palette = {
"Tumor": "#F4AD2A",
"Normal Macrophage": "#76A3D0", # white violin (no fill)
"TAM": "#FF5835"
}
[52]:
selected_gene = "MYC"
df = pd.DataFrame({
"group": adata_plot.obs["celltype"].astype(str),
"value": adata_plot[:, selected_gene].X.flatten()
})
plot_order = ["Normal Macrophage", "TAM", "Tumor"]
# Plot
plt.figure(figsize=(5, 5))
ax = sns.violinplot(
x="group",
y="value",
data=df,
order=plot_order,
scale="width",
inner=None,
linewidth=1.5,
palette=palette,
bw=0.3
)
# Overlay dot strip
sns.stripplot(
x="group",
y="value",
order=plot_order,
data=df,
color="black",
size=2,
jitter=0.25,
alpha=0.8
)
# Log scale y-axis
ax.set_yscale("log", base=2)
ax.set_ylim(bottom=1) # Set y-axis limits to avoid log scale issues
# ax.set_yticks([1, 3, 10, 30])
# ax.set_yticklabels(["1", "3", "10", "30"], fontsize=12)
# Customize spines and ticks
ax.spines['top'].set_visible(False)
ax.spines['right'].set_visible(False)
ax.tick_params(axis='x', labelsize=14)
ax.set_xlabel("")
ax.set_ylabel("")
# Bold gene label title
plt.title("")
plt.tight_layout()
plt.show()
/tmp/ipykernel_1057144/3781531446.py:11: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
/tmp/ipykernel_1057144/3781531446.py:11: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
/tmp/ipykernel_1057144/3781531446.py:11: FutureWarning:
The `bw` parameter is deprecated in favor of `bw_method`/`bw_adjust`.
Setting `bw_method=0.3`, but please see docs for the new parameters
and update your code. This will become an error in seaborn v0.15.0.
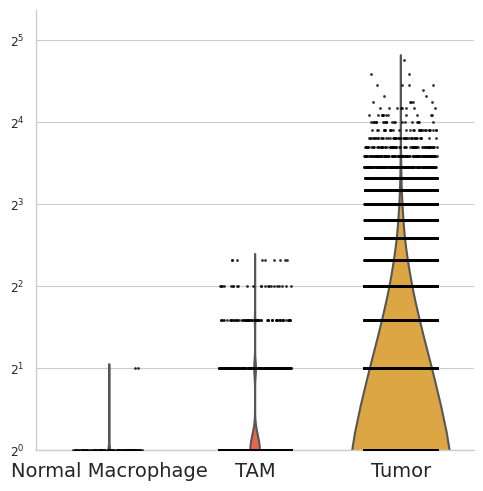
[53]:
selected_gene = "MMP12"
df = pd.DataFrame({
"group": adata_plot.obs["celltype"].astype(str),
"value": adata_plot[:, selected_gene].X.flatten()
})
plot_order = ["Normal Macrophage", "TAM", "Tumor"]
# Plot
plt.figure(figsize=(5, 5))
ax = sns.violinplot(
x="group",
y="value",
data=df,
order=plot_order,
scale="width",
inner=None,
linewidth=1.5,
palette=palette,
bw=0.3
)
# Overlay dot strip
sns.stripplot(
x="group",
y="value",
order=plot_order,
data=df,
color="black",
size=2,
jitter=0.25,
alpha=0.8
)
# Log scale y-axis
ax.set_yscale("log", base=2)
ax.set_ylim(bottom=1) # Set y-axis limits to avoid log scale issues
# ax.set_yticks([1, 3, 10, 30])
# ax.set_yticklabels(["1", "3", "10", "30"], fontsize=12)
# Customize spines and ticks
ax.spines['top'].set_visible(False)
ax.spines['right'].set_visible(False)
ax.tick_params(axis='x', labelsize=14)
ax.set_xlabel("")
ax.set_ylabel("")
# Bold gene label title
plt.title("")
plt.tight_layout()
plt.show()
/tmp/ipykernel_1057144/639527426.py:11: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
/tmp/ipykernel_1057144/639527426.py:11: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
/tmp/ipykernel_1057144/639527426.py:11: FutureWarning:
The `bw` parameter is deprecated in favor of `bw_method`/`bw_adjust`.
Setting `bw_method=0.3`, but please see docs for the new parameters
and update your code. This will become an error in seaborn v0.15.0.
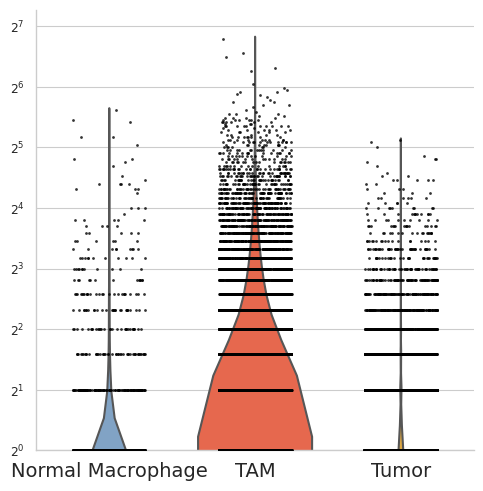
[37]:
selected_gene = "SPP1"
df = pd.DataFrame({
"group": adata_plot.obs["celltype"].astype(str),
"value": adata_plot[:, selected_gene].X.flatten()
})
plot_order = ["Normal Macrophage", "TAM", "Tumor"]
# Plot
plt.figure(figsize=(5, 5))
ax = sns.violinplot(
x="group",
y="value",
data=df,
order=plot_order,
scale="width",
inner=None,
linewidth=1.5,
palette=palette,
bw=0.3
)
# Overlay dot strip
sns.stripplot(
x="group",
y="value",
order=plot_order,
data=df,
color="black",
size=2,
jitter=0.25,
alpha=0.8
)
# Log scale y-axis
ax.set_yscale("log", base=2)
ax.set_ylim(bottom=1) # Set y-axis limits to avoid log scale issues
# ax.set_yticks([1, 3, 10, 30])
# ax.set_yticklabels(["1", "3", "10", "30"], fontsize=12)
# Customize spines and ticks
ax.spines['top'].set_visible(False)
ax.spines['right'].set_visible(False)
ax.tick_params(axis='x', labelsize=14)
ax.set_xlabel("")
ax.set_ylabel("")
# Bold gene label title
plt.title("")
plt.tight_layout()
plt.show()
/tmp/ipykernel_1057144/1361498247.py:11: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
/tmp/ipykernel_1057144/1361498247.py:11: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
/tmp/ipykernel_1057144/1361498247.py:11: FutureWarning:
The `bw` parameter is deprecated in favor of `bw_method`/`bw_adjust`.
Setting `bw_method=0.3`, but please see docs for the new parameters
and update your code. This will become an error in seaborn v0.15.0.
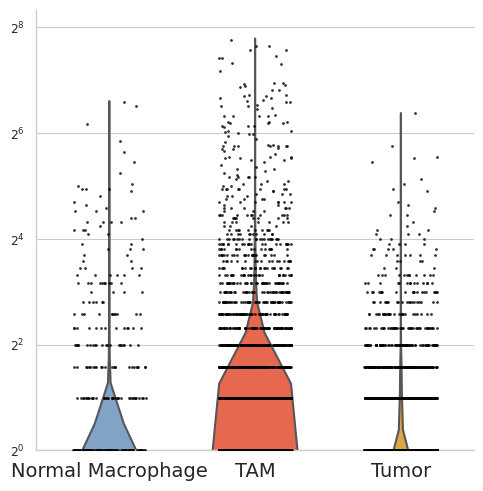
Legend receptor
[9]:
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
import cmasher as cmr
def lr_heatmap_mag_target(lr_res, sources, target, filter_thres1s, filter_thres2s, n_top_orders,
source_names=None, target_name=None,
add_lrs=None, cmap_lr=None, leg_bbox=None, mx=None, my=None, rot_x=90):
order_metric = 'magnitude_rank'
filter_metric1 = 'magnitude_rank'
filter_metric2 = 'magnitude_rank'
keep_cols = ['lr', 'source', 'target', 'magnitude_rank']
dfs = []
for i, source in enumerate(sources):
df = lr_res.loc[(lr_res.source==source)&(lr_res.target==target)]
df0 = df.loc[(df[filter_metric1]<=filter_thres1s[i])&(df[filter_metric2]<=filter_thres2s[i])]
df_ = df0[keep_cols].sort_values(order_metric).head(n_top_orders[i])
lrs_ = df_.lr.values.tolist()
df_ = lr_res.loc[(lr_res.lr.isin(lrs_))&(lr_res.source.isin(sources))&(lr_res.target==target)][keep_cols]
print(source, len(lrs_), df_.shape)
if add_lrs is not None:
for lr in add_lrs:
if lr not in df_.lr.values:
if lr in df.lr.values:
df_ = pd.concat([df_, df.loc[df.lr==lr][keep_cols]])
else:
df_ = pd.concat([df_, pd.DataFrame([[lr, source, target, 1, 1]], columns=keep_cols)])
df_ = df_.sort_values(order_metric)
dfs.append(df_)
concat_df = pd.concat(dfs).drop_duplicates()
print(len(concat_df), concat_df.lr.unique().shape)
spec_long = concat_df[['lr', 'source', 'magnitude_rank']]
spec_wide = spec_long.pivot(index='lr', columns='source', values='magnitude_rank').fillna(1)
spec_wide['min'] = spec_wide.min(1)
spec_wide = spec_wide.sort_values('min')
spec_wide = spec_wide.iloc[:,:len(sources)]
mag_long = concat_df[['lr', 'source', 'magnitude_rank']]
mag_wide = mag_long.pivot(index='lr', columns='source', values='magnitude_rank').fillna(1)
mag_wide['min'] = mag_wide.min(1)
mag_wide = mag_wide.sort_values('min')
mag_wide = mag_wide.iloc[:,:len(sources)]
print('Combine:', len(mag_wide))
mag_df = mag_wide.stack().reset_index(name='mag')
spec_df = spec_wide.stack().reset_index(name='spec')
mag_spec_df = mag_df.merge(spec_df, on=['lr', 'source'])
mag_spec_df['mag'] = -np.log10(mag_spec_df['mag'].values)
mag_spec_df['spec'] = -np.log10(mag_spec_df['spec'].values)
mag_spec_df['source'] = pd.Categorical(mag_spec_df['source'], categories=sources)
## heatmap
plt.rcParams.update({"figure.dpi": 100})
sns.set_theme(style="whitegrid")
sns.set_context('paper',font_scale=1.)
# cmap_lr = cmr.get_sub_cmap(mpl.cm.magma, 0.05,0.95)
if cmap_lr is None:
cmap_lr = cmr.get_sub_cmap(sns.cubehelix_palette(rot=0.1, dark=0.2, light=0.8, as_cmap=True, reverse=True), 0, 1)
if source_names is None:
source_names = sources
xticklabels = source_names
fs = 10
w = 3
h = 0.3 * len(mag_wide)
if mx is None or my is None:
mx = 0.5/(len(sources)-1) if len(sources) > 1 else 5
my = 0.5/(len(mag_wide)-1)
sizes = (10, 250)
hue_max = mag_spec_df['spec'].max()
size_max = mag_spec_df['mag'].max()
g = sns.relplot(
data=mag_spec_df,
x="source", y="lr", hue="spec", size="mag",
palette=cmap_lr, hue_norm=(0,hue_max), size_norm=(0,size_max), edgecolor=".7",
height=h, aspect=1, sizes=sizes, legend=True,
)
if target_name is None:
target_name = target
title = f'$\\rightarrow$ {target_name}'
g.set(xlabel="", ylabel="", xticklabels=xticklabels, title=None, aspect='equal')
g._axes[0][0].set_title(title, fontsize=fs+2, color='darkblue')
g.set_xticklabels(size = fs, rotation=rot_x)
g.set_yticklabels(size = fs, style='italic')
g.despine(left=True, bottom=True)
g.ax.margins(x=mx, y=my)
if leg_bbox is not None:
for i in range(len(g._legend.texts)):
if g._legend.texts[i].get_text() == 'spec':
g._legend.texts[i].set_text('Spec.')
if g._legend.texts[i].get_text() == 'mag':
g._legend.texts[i].set_text('Mag.')
sns.move_legend(
g, "center", bbox_to_anchor=leg_bbox, fontsize=fs, ncols=1, frameon=False, alignment='left',
)
else:
g._legend.remove()
return concat_df
import liana as li
adata = adata_boundary.copy()
[10]:
expr_prop = 0.05
lrTAM = li.mt.rank_aggregate(adata,
groupby='celltype',
resource_name='consensus',
expr_prop=expr_prop,
use_raw=False,
verbose=True,
inplace=False)
Using resource `consensus`.
Using `.X`!
Converting to sparse csr matrix!
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/anndata/_core/anndata.py:522: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
74 samples of mat are empty, they will be removed.
Make sure that normalized counts are passed!
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/liana/method/_pipe_utils/_pre.py:153: FutureWarning: The default of observed=False is deprecated and will be changed to True in a future version of pandas. Pass observed=False to retain current behavior or observed=True to adopt the future default and silence this warning.
0.59 of entities in the resource are missing from the data.
Generating ligand-receptor stats for 94616 samples and 568 features
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/liana/method/sc/_liana_pipe.py:262: ImplicitModificationWarning: Setting element `.layers['scaled']` of view, initializing view as actual.
Assuming that counts were `natural` log-normalized!
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/liana/method/sc/_liana_pipe.py:360: RuntimeWarning: overflow encountered in power
Running CellPhoneDB
100%|██████████████████████████████████████████████████████████████████████████████| 1000/1000 [00:58<00:00, 16.98it/s]
Running Connectome
Running log2FC
Running NATMI
Running SingleCellSignalR
[11]:
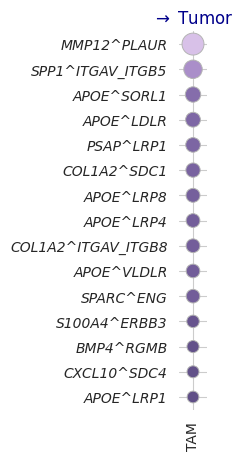
lrTAM['lr'] = lrTAM[['ligand_complex', 'receptor_complex']].agg('^'.join, axis=1)
# Mapping source / target names: Macrophage -> TAM, Tumor -> Tumor, CD8 Cytotoxic T cell -> CD8 T cell
lrTAM.source = lrTAM.source.replace({'Macrophage': 'TAM'})
lrTAM.target = lrTAM.target.replace({'CD8 Cytotoxic T cell': 'CD8 T cell'})
lrTAM.target = lrTAM.target.replace({'Tumor III': 'Tumor'})
# Exclude: source is TAM and lr starts with COL1A1 (cancer associated fibroblast ligands, due to diffusion)
lrTAM = lrTAM[~((lrTAM.source == 'TAM') & (lrTAM.ligand_complex.str.startswith('COL1A1')))]
temp = lr_heatmap_mag_target(lrTAM,
sources=['TAM'],
target='Tumor',
filter_thres1s=[1,1],
filter_thres2s=[1,1],
n_top_orders=[15]*3,
leg_bbox=None)
TAM 15 (15, 4)
15 (15,)
Combine: 15
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/seaborn/axisgrid.py:123: UserWarning: The figure layout has changed to tight
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/seaborn/axisgrid.py:44: UserWarning: FixedFormatter should only be used together with FixedLocator
[12]:
raw_ct = adata_svt.obs["celltype"].copy().astype(str)
# TAM index
tam_idx = adata[adata.obs["celltype"].isin(["Macrophage"])].obs_names
raw_ct[tam_idx] = "TAM"
adata_svt.obs["celltype"] = raw_ct.astype("category")
[15]:
mapping_dict = {
"Tumor_III PLAUR": "#F4AD2A", # ""SDC1"
"TAM MMP12": "#FF5835",
}
temp = plot_specific_celltype_gene(adata_svt, mapping_dict, x_col = "x", y_col = "y", s = 0.5, revert_y = False, revert_x= False)

[ ]:
mapping_dict = {
"Tumor_III ITGAV": "#F4AD2A",
"Tumor_III ITGB5": "#F4AD2A",
"TAM SPP1": "#FF5835",
}
temp = plot_specific_celltype_gene(adata_svt, mapping_dict, x_col = "x", y_col = "y", s = 0.5, revert_y = False, revert_x= False)

[16]:
mapping_dict = {
"Tumor_III SORL1": "#F4AD2A",
"TAM APOE": "#FF5835",
}
temp = plot_specific_celltype_gene(adata_svt, mapping_dict, x_col = "x", y_col = "y", s = 0.5, revert_y = False, revert_x= False)

Visualization for figure 5 g
[42]:
mapping_dict = {
"Macrophage SPP1": "#FF0000",
"Tumor_III all": "grey",
}
plot_dict_1 = plot_specific_celltype_gene(adata_svt, mapping_dict, x_col = "x", y_col = "y", s = 0.5, revert_y = False, revert_x= False)
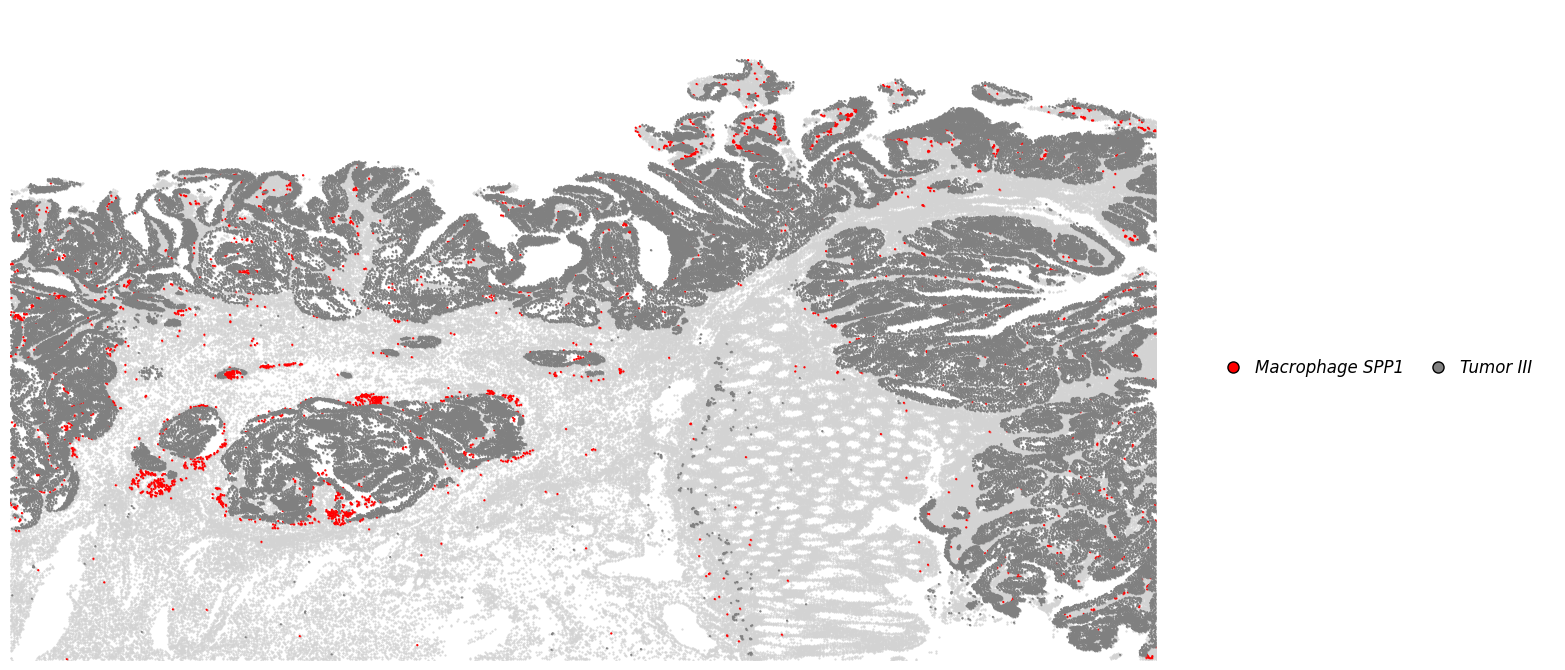
[43]:
mapping_dict = {
"Macrophage SELENOP": "#BC05D8",
"Tumor_III all": "grey",
}
plot_dict_2 = plot_specific_celltype_gene(adata_svt, mapping_dict, x_col = "x", y_col = "y", s = 0.5, revert_y = False, revert_x= False)
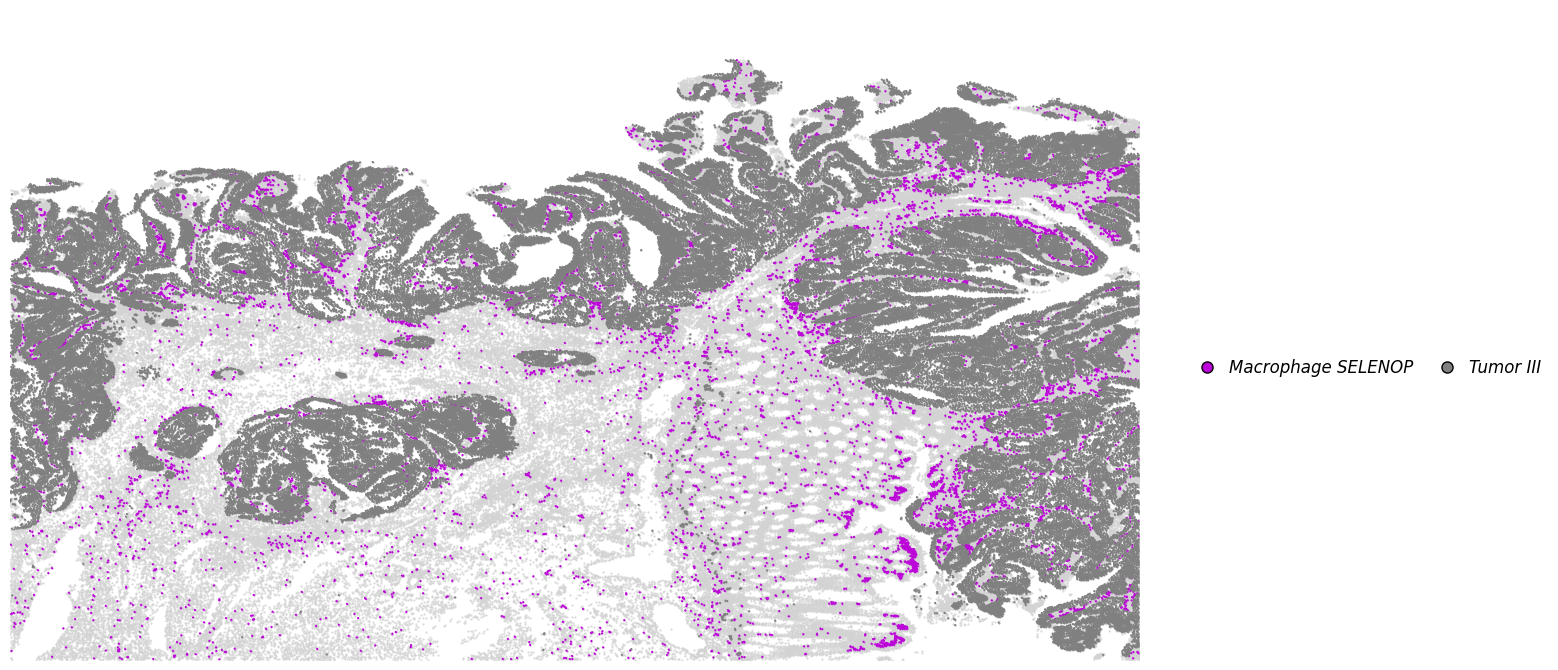
[44]:
adata_spp1 = plot_dict_1["Macrophage SPP1"].copy()
adata_spp1.obs["celltype"] = "Macrophage SPP1+"
c1 = adata_spp1[(adata_spp1.obs["x"] > 500) & (adata_spp1.obs["x"] < 1500) & (adata_spp1.obs["y"] < 1500) & (adata_spp1.obs["y"] > 300)].obs_names # .loc[:, "celltype"]= "Marophage SPP1+ Cluster 1"
c2 = adata_spp1[(adata_spp1.obs["x"] > 2400) & (adata_spp1.obs["x"] < 3500) & (adata_spp1.obs["y"] < 2500) & (adata_spp1.obs["y"] > 1500)].obs_names
adata_spp1.obs.loc[c1, "celltype"] = "Marophage SPP1+ Cluster 1"
adata_spp1.obs.loc[c2, "celltype"] = "Marophage SPP1+ Cluster 2"
adata_selenop = plot_dict_2["Macrophage SELENOP"].copy()
adata_selenop.obs["celltype"] = "Macrophage SELENOP+"
temp = anndata.concat([adata_spp1, adata_selenop])
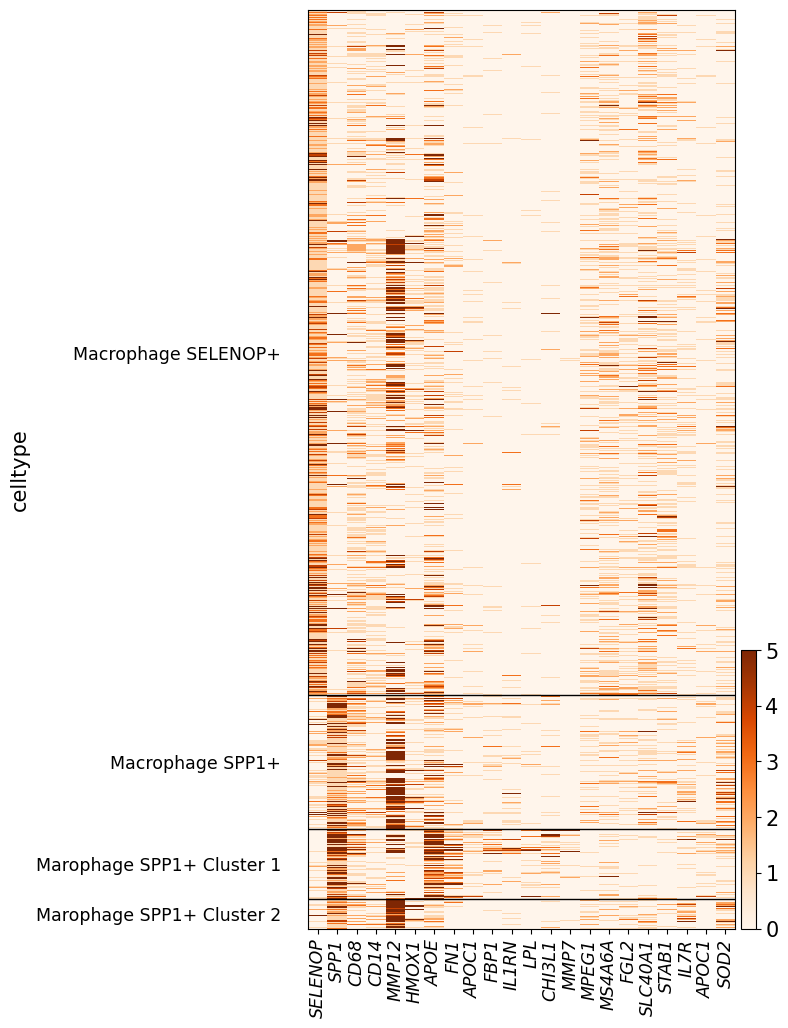
selected_gene = [
"SELENOP", "SPP1", "CD68", "CD14", "MMP12", "HMOX1", "APOE", "FN1", "APOC1", "FBP1", "IL1RN", "LPL", "CHI3L1", "MMP7",
"MPEG1", "MS4A6A", "FGL2", "SLC40A1", "STAB1", "IL7R", "APOC1", "SOD2"
]
gene_list = selected_gene
# As type categorical
temp.obs['celltype'] = temp.obs['celltype'].astype('category')
# All set to white
temp.uns["celltype_colors"] = ["#FFFFFF"] * len(np.unique(adata_plot.obs['celltype']))
# Only show invasive tumor and Prolif Invasive Tumor
with plt.rc_context({'font.size': 15}):
hm = sc.pl.heatmap(temp, gene_list, groupby='celltype', cmap='Oranges', show_gene_labels=True, vmax=5, swap_axes=False, figsize=(6,12), show=False)
# Delete groupby_ax
ax = hm['heatmap_ax']
for l in ax.get_xticklabels():
l.set_style("italic")
# Show with high dpi
plt.show(block=True)
/home/yhchenmath/anaconda3/envs/cellseg/lib/python3.9/site-packages/anndata/_core/anndata.py:1906: UserWarning: Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
utils.warn_names_duplicates("obs")